Article Text

Abstract
Introduction Neoadjuvant chemotherapy (NACT) is an emerging approach for locally advanced cervical cancer (LACC). However, the clinical response and postoperative adjuvant radiation or chemoradiation trimodality treatment resulted in controversy. PD-1 inhibitors have shown promising role in recurrent or metastatic cervical cancer, and there is preclinical evidence of the activation and synergistic effects of NACT on PD-1 inhibitors. This study aims to evaluate the efficacy and safety of the preoperative PD-1 inhibitor camrelizumab combined with NACT for LACC.
Methods and analysis The study is designed as a multicentre, open-label, single-arm, prospective phase II study. A total of 82 patients will receive neoadjuvant chemo-immunotherapy, defined as one cycle of cisplatin (75–80 mg/m2, intravenously) plus nab-paclitaxel (260 mg/m2, intravenously) NACT and subsequent two cycles of camrelizumab (200 mg, intravenously) combined with NACT. After neoadjuvant chemo-immunotherapy, patients exhibiting complete response and partial response will undergo radical surgery and subsequent adjuvant therapy. In contrast, patients with stable disease and progressive disease will transfer to concurrent chemoradiotherapy (CCRT). Following surgery, patients will receive adjuvant CCRT or radiotherapy. The primary endpoint is the objective response rate. The secondary endpoints are the pathological complete response, patients requiring postoperative adjuvant therapy, safety of neoadjuvant chemo-immunotherapy, surgical complication, event-free survival, and overall survival. An additional aim is to dynamically evaluate peripheral immune responses and local immunological microenvironments and their association with neoadjuvant immunotherapy.
Ethics and dissemination This trial was approved by the Medical Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (S2020-112). This study is among the first to evaluate the efficacy and safety of neoadjuvant chemo-immunotherapy in LACC. The findings of this research will promote neoadjuvant anti-PD-1 immunotherapy with radical surgery as a new therapeutic strategy.
Trial registration number ClinicalTrials.gov Registry (NCT04516616).
- Clinical trials
- Gynaecological oncology
- IMMUNOLOGY
- CHEMOTHERAPY
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This is a multicentre, prospective cohort study to evaluate the efficacy and safety of neoadjuvant chemo-immunotherapy in locally advanced cervical cancer.
Priming chemotherapy will be used before anti-PD-1-combined chemotherapy.
Paclitaxel is replaced with nab-paclitaxel as a chemotherapy regimen, avoiding the combined use of glucocorticoids.
Blood samples and tumour tissue biopsies will be collected into a translational research programme.
Since this study is a single-arm study, it may include some bias.
Introduction
Cervical cancer is the fourth most frequently diagnosed cancer and the fourth most common cause of cancer-related deaths among women worldwide, accounting for approximately 604 000 new cases and 342 000 deaths in 2020.1 Locally advanced cervical cancer (LACC) includes patients with International Federation of Gynecology and Obstetrics (FIGO) stage IB3–IVA disease, for whom concurrent chemoradiotherapy (CCRT) is the standard treatment. However, definitive surgery can also be performed in patients with stage IB3 or IIA disease.2 3 Characterised by the large extent of malignant tissue, it is very difficult to complete surgical resection. Furthermore, the prognosis of LACC has not improved significantly over the past decades, with an average 5-year survival rate of 60%–82.3%.4–6 Hence, treatment options that improve survival in this subgroup of patients remained as a major public health concern, and effective therapies for further improvement in survival must be developed.
In areas where radiotherapy apparatus is not available, platinum-based neoadjuvant chemotherapy (NACT) followed by radical hysterectomy has been proposed as an alternative approach. This alternative approach increases the opportunities for radical surgery and indicates a similar prognosis compared with CCRT. The NACT, however, is effective in only two-thirds of patients, and those who do not respond receive little benefit from it.7 8 Moreover, 32.2% of patients required postoperative adjuvant radiation or chemoradiation, resulting in controversy in terms of health economics and trimodality treatment.5 Therefore, although NACT has been widely used in many countries, it is not recommended as a first-line treatment for LACC.
Recently, immune checkpoint inhibitors (ICIs) have made remarkable advances in immunotherapy for cervical cancer. Additionally, pembrolizumab combined with chemotherapy has been approved as a first-line treatment for advanced PD-L1-positive cervical cancer.9 10 Previous clinical trials have shown that immunotherapy, with a favourable toxicity profile and durable response, has made a tremendous breakthrough in the treatment of cervical cancer. Contrarily, immuno-monotherapy shows a relatively low response rate. In advanced or recurrent cervical cancer, ICI monotherapy has a response rate of approximately 15%, and combination therapy with chemotherapy, radiotherapy or other immune-targeted therapies has improved response rates of 50%–60%.11 12
Neoadjuvant chemo-immunotherapy has attained milestones in the treatment of many solid tumours. In contrast, most cervical cancer researches have focused on immunotherapy in patients with metastatic or recurrent disease, with very few studies exploring neoadjuvant immunotherapy in LACC. Recent theoretical developments have revealed that chemotherapeutic agents possess immunomodulatory properties, modulating immune cells and shaping their functions. Conventional chemotherapeutics have been known to promote immunogenicity of the tumour because of the modulation of the anti-tumour T cell response through triggering the release of tumour-derived antigens, inducing immunogenic cell death, disrupting the immunosuppressive microenvironment and enhancing the effector T cell response.13 In addition, clinical research supports that NACT can increase PD-L1 expression in cervical cancer, and overexpression of PD-L1 is significantly associated with a better response to ICIs, such as the PD-1/PD-L1 blockade.14 These findings show that reprogramming the tumour immunological microenvironment using NACT may potentially sensitise cervical tumours to immunotherapy and act synergistically with ICIs, similar to early triple-negative breast cancer (TNBC) and non-small cell lung cancer (NSCLC).15 16
Camrelizumab is a human IgG4 monoclonal antibody that targets the PD-1 immune checkpoint receptor. Favourable efficacy and tolerable safety profiles have been demonstrated in several advanced tumours. We perform this pilot study to evaluate the clinical activity and safety of NACT combined with PD-1 inhibitors in patients with LACC. Furthermore, we will explore the relationship between the tumour microenvironment landscape and response to therapy, as well as dynamic changes in intratumoral and peripheral immune microenvironments.
Methods and analysis
Trial design
The study is designed as a multicentre, open-label, single-arm, prospective phase II study. Eighty-two patients will receive neoadjuvant immunotherapy, defined as one cycle of cisplatin plus nab-paclitaxel NACT and subsequent two cycles of PD-1 antibody combined with NACT. Figure 1 shows the flow chart of the trial. Patients will be recruited from large 3A hospitals, and a list of all participating centres is in online supplemental table 1. The patient registration process began from December 2020 to December 2022. The 5-year follow-up is designed for all patients, and the final study report will be prepared within 6 months. Therefore, this study is scheduled to end in July 2028.
Supplemental material

The overall trial flow of the study. 1If there are contraindications for MRI, CT is recommended. 2In priming phase and NACIT phases, blood samples are collected before (~24 hours) and after (~24 hours) each cycle; in patients undergoing surgery, blood samples are collected before and then 7 days after radical surgery, if any, after administration of postoperative adjuvant treatment; in patients undergoing CCRT, blood samples are collected before and immediately after CCRT. 3In priming phase and NACIT phases, biopsy samples of tumour tissue are collected before each cycle; in patients undergoing surgery, the resected tumour tissue and a paracancer normal tissue will be collected; in patients undergoing CCRT, biopsy sample is collected before CCRT. CCRT, concurrent chemoradiotherapy; CPS, combined positive score; CR, complete response; D1, day 1; D2, day 2; FIGO, International Federation of Gynecology and Obstetrics; MRI, magnetic resonance imaging; NACIT, neoadjuvant chemo-immunotherapy; NCCN, National Comprehensive Cancer Network; PD, progressive disease; PR, partial response; Q3M, every 3 months; Q6M, every 6 months; SD, stable disease.
The objective of the study
This study aims to evaluate the effects of neoadjuvant chemo-immunotherapy on tumour response (including clinical response and pathological response), the proportion of patients requiring postoperative adjuvant therapy, treatment-related toxicity, subsequent surgical complications and survival (including event-free survival (EFS) and overall survival (OS)) of patients with LACC. An additional aim of this study is to dynamically evaluate peripheral immune responses and local immunological microenvironments, and their association with neoadjuvant immunotherapy efficacy.
Endpoints of the study
The primary endpoint is the objective response rate (ORR), defined as the percentage of the participants who achieve a complete response (CR) or partial response (PR). The primary endpoint analysis will include participants who receive camrelizumab therapy and have at least one post-baseline tumour assessment. The ORR will be assessed by a blind independent central reviewer per Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST 1.1). The secondary endpoints are the rate of pathological complete response (pCR), the number of patients with treatment-related adverse events (AEs), serious AEs or immune-related AEs (irAEs), the number of patients with surgical complications and postoperative adjuvant therapy, EFS and OS. Histological examination of the entire resected specimen will be performed to identify patients with pCR, defined as the absence of viable tumour cells on all slides. The optimal response, based on the Buda criteria, is a complete disappearance of the tumour in the cervix with negative nodes or a residual disease with less than 3 mm stromal invasion, including in situ carcinoma.17 The Common Terminology Criteria for Adverse Events (V.5.0) is used to evaluate AEs after the first dose of chemotherapeutic drugs to 30 days after the last dose. The safety set used for the safety evaluation will include patients using camrelizumab at least once and with relatively complete medical records. Postoperative complication is defined as any clinically significant deviation from a normal postoperative course. EFS is defined as the time interval from the date of enrollment to disease progression, local or distant recurrence (in patients undergoing surgery), or death due to any cause. OS is defined as the date of enrollment until the date of death due to any cause or the last follow-up visit. Additionally, in participants who undergo radical surgery, disease-free survival (DFS) is calculated separately from the date of completing prescribed treatment to the date of disease relapse or death, irrespective of the cause.
Study procedures
Patient selection/screening
Eligible patients are 18–70 years of age and have newly diagnosed cervical cancer of stage IB3, IIA2 or IIB/IIIC1r with a tumour diameter ≥4 cm (FIGO, 2018). All patients will be PD-L1 positive, with pathologically confirmed squamous cell carcinoma, adenocarcinoma or adenosquamous carcinoma, and have normal organ function. The detailed inclusion and exclusion criteria are listed in table 1. Informed consent will be obtained before PD-L1 immunohistochemistry assays and the participant will receive one cycle of cisplatin and nab-paclitaxel treatment. PD-L1 expression is evaluated using the 22C3 antibody clone (Agilent, Dako). After randomisation, participants who do not satisfy the inclusion or exclusion criteria will be excluded. From published data, 30%~88% of patients with cervical cancer are PD-L1 positive,18 19 and it is feasible that patients who turned out PD-L1 negative will be excluded.
Inclusion and exclusion criteria
Treatment
Patients enrolled in this cohort will first receive one cycle of platinum-based doublet NACT. The regimens are cisplatin 75–80 mg/m2 and nab-paclitaxel 260 mg/m2, to be administered intravenously. After 3 weeks, participants will receive two cycles of PD-1 inhibitor combined with NACT once every 3 weeks. The regimens are cisplatin 75–80 mg/m2 intravenously plus nab-paclitaxel 260 mg/m2 intravenously on day 1 and camrelizumab 200 mg intravenously on day 2. Tumour response will be assessed by MRI using RECIST 1.1 criteria 3 weeks after the last dose of neoadjuvant therapy. If patients achieve CR or PR, an open laparotomy radical hysterectomy plus pelvic lymphadenectomy will be performed, with or without para-aortic lymphadenectomy. Following surgery, patients will receive therapy as the National Comprehensive Cancer Network (NCCN) guidelines recommended. Patients with high-risk factors (pelvic lymph node positive, margin positive or parametrial infiltration) should be treated with cisplatin-based CCRT. Conversely, those with intermediate risk factors as in the Sedlis criteria will receive adjuvant radiotherapy. However, patients with stable disease (SD) or tumour progression (PD) will receive concurrent CCRT. In the case of grade 3–4 AEs, the treatment should be suspended, and the AEs should be actively treated until they return to grades 1–2. The dose of cisplatin and nab-paclitaxel may be reduced in the treatment in case of serious AE. However, discontinuation of camrelizumab is recommended if irAEs do not recover to grade 2 or lower before starting the second cycle of combined therapy.
Data collection procedure
Informed consent forms will be obtained from each participant before the screening evaluations. Baseline data will be recorded at the screening visit and completed within 28 days before enrolment. In addition, data will be collected at baseline, during neoadjuvant immunotherapy, surgery, postoperative adjuvant therapy or CCRT. Online supplemental table 2 provides an overview of the collection of the outcomes.
Follow-up
After the initially recommended therapy is completed, routine follow-up will be scheduled every 3 months for 1 year and every 6 months after that. Patients will be followed up for at least 5 years. Follow-up includes pelvic examinations, imaging examinations, serum tumour markers and cytology. Imaging examinations include abdominal and pelvic CT or MRI, and abdominal imaging is allowed to perform every 6 months in the first year. Vaginal stump brushing cytology and human papillomavirus testing are performed simultaneously. The serum tumour markers are squamous cell carcinoma antigen for patients with squamous cell carcinoma, CA125 for adenocarcinoma and both for adenosquamous carcinoma. Systematic examination revealed tumour recurrence would result in whole-body restaging. Further individual examination, post-recurrence anti-tumour therapy and survival follow-up need to be performed.
Translational research
Blood samples are obtained from patients before initiation of and after every cycle of chemotherapy and cervical cancer biopsy samples are additionally obtained immediately before every cycle of therapy. The blood samples of patients undergoing radical surgery are collected before and 7 days after the surgery, if any, after administering postoperative adjuvant treatment. Additionally, surgically resected tumour tissues and adjacent normal tissues will be collected. On the other hand, blood and cervical cancer biopsy samples will be taken before CCRT, and blood samples will be taken again at the end of the therapy in patients with SD/PD who transfer to CCRT. The detailed sample collection process is presented in figure 1.
All the patients participating in the trial may be included in a translational research programme as stated in the informed consent. The analysis of the predictive value of available immune microenvironmental factors in responders, possible therapy-associated changes in different tumour regions, tumour microenvironment and peripheral blood are among the major interests. Detailed research will be conducted to further investigate the immunotherapy reaction with tumour gene mutations and defects, the tumour immune microenvironment and mesenchymal cells. Dynamic panoramas of intratumoral infiltrating immune cells and peripheral blood will be performed with multiple omics research.
Statistical analysis
Sample size calculation
The clinical response rate in patients with LACC after NACT is approximately 70%.20 21 With a one-sided binomial test at an overall type I error rate of a=0.05, a sample size of 69 patients is needed to maintain 91.4% power under the hypothesis that the overall response rate of neoadjuvant immunotherapy is expected to increase to 85%. Eighty-two patients are included in the study, with an estimated dropout rate of 15%.
Data analyses
The efficacy analysis will include participants who receive camrelizumab and have at least one post-baseline tumour assessment. The participants undergoing surgery will be evaluated for surgical pathological findings and any operative complications. CIs will be estimated for ORR and pathological response rate using the Clopper-Pearson method. EFS, DFS and OS analysis will be performed by the Kaplan-Meier curve, providing the median time to the event, and 95% CIs are calculated using the Greenwood formula. The enrolled patients who receive at least one dose of camrelizumab will be included in the safety analysis. In general, descriptive measures are used to summarise continuous variables (average, SD, median, maximum and minimum values). Categorical variables are expressed as frequencies and percentages. Cox univariate and multivariate analyses are used to analyse the correlation between biomarker expression, clinical efficacy and prognosis for treatment sensitivity analysis of the biomarkers. All data collected on the case report form (CRF) will be listed per patient. Any deviation from the statistical methods given in the protocol will be reported in the final report as appropriate. Outcomes with p<0.05 are considered statistically significant. Furthermore, no interim analysis of the data will be conducted.
Data management and monitoring
Detailed data obtained from the patients will be recorded in the CRF and a central database is responsible for data collection and management. All study records and original documents will be maintained and stored according to relevant regulations and guidelines, or by the research institution’s rules. The investigator accessing the relevant raw data of the clinical study is responsible for reviewing the CRF to determine the information’s completeness, accuracy and consistency with the source data. A trained and qualified central monitoring group will periodically visit each participating centre throughout the trial to ensure data submission, patient eligibility and protocol compliance.
Patient and public involvement
Patients or the public are not involved in this clinical trial’s design, conduct or reporting.
Ethics and dissemination
The Medical Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology, China approved the study on 10 June 2020 (approval number: S2020-112). An amendment was approved on 30 November 2022 (approval number: S2020-112-3). The protocol of this study has been registered at ClinicalTrials.gov (identification number: NCT04516616). Participants who suffer harm from trial participation will receive compensation from the insurance company. As the study is still in progress, results will be published when the last enrolled patient finishes the primary endpoint evaluation, and finishes completion of 3-year and 5-year follow-ups. The findings from the analysis of the trial will be disseminated through scientific and professional conferences, and published in peer-reviewed journals.
Discussion
This study is one of the first multicentre, prospective cohort studies to evaluate the efficacy and safety of neoadjuvant chemo-immunotherapy in LACC. The results of this research will provide the foundation for developing alternative therapeutic strategies for LACC, and provide molecular biology of NACT and immunotherapy on the intratumoral and peripheral immune microenvironments in patients with cervical cancer. Although several similar clinical trials have been carried out since we registered the protocol, the unique characteristic of this clinical trial is that participants will receive upfront priming chemotherapy followed by PD-1 inhibitor combined with chemotherapy.
Although CCRT is the standard primary treatment for LACC, several challenges remain to be overcome. Difficulties exist in implementing standard radiation schedules in low/middle-income countries because of the high cost of radiotherapy facilities. Additionally, patients would experience serious side effects, including gastrointestinal toxicity, sexual dysfunction and fertility concerns in young patients, severely affecting their quality of life.22 23 Next, radiation-induced pelvic fibrosis increases the surgical difficulty and risk, and in case of progression or recurrence after radiation, treatment options are extremely limited. Finally, radio-resistant cervical cancer has a high potential for recurrence, including regional and distant metastasis; thus, early initiation of systemic therapy is imperative to further improve survival. Over the past two decades, NACT combined with surgery has been the preferred treatment for LACC in low/middle-income countries that lack radiotherapy equipment. NACT can effectively reduce the size of bulky tumours and improve the chances of performing radical surgery. However, survival benefits have only been demonstrated in patients with clinical responses.24 In addition, more than one-third of patients treated with NACT and surgery require postoperative radiotherapy, increasing the risk of treatment-related complications.25 Therefore, therapeutic options that improve clinical response and avoid postoperative adjuvant radiotherapy based on reduced surgical pathological findings are warranted.
Since interleukin-2, the first immunotherapy approved for metastatic melanoma in 1998,26 the promising role of immunotherapy in solid tumours has gained increasing attention, especially in recurrent or metastatic disease. The Food and Drug Administration has approved pembrolizumab, a PD-1 inhibitory antibody for cervical cancer, following the outcome of the KEYNOTE-158 clinical trial.10 A series of clinical trials have demonstrated the clinical benefits of ICIs in patients with recurrent or metastatic cervical cancer, showing promising overall response rates and well-tolerated toxicity profiles.27 28 Immunotherapy is recommended as the initial therapy for several solid tumours, apart from second-line treatment in recurrent/metastatic cases. with the new progress in the molecular mechanism of cancer treatment. Recent publications indicated that ICIs-based neoadjuvant therapy was superior to chemotherapy alone in NSCLC, with reported major pathological response rates of 20%–85%.29–31 Moreover, studies have shown that neoadjuvant immunotherapy in combination with chemotherapy improves the pCR and EFS rates in early TNBC. The KEYNOTE-522 Study showed that the pembrolizumab-chemotherapy group had significantly higher EFS (84.5%) at 36 months than the chemotherapy group (76.8%).32 Pembrolizumab has been recommended as a preoperative neoadjuvant and postoperative adjuvant combination treatment for TNBC in several countries based on the results of this study.
However, ICI monotherapy does not maximise the benefits; researchers have also explored the optimised neoadjuvant therapy scheme of ICIs combined with chemotherapy. PD-1 inhibitor monotherapy has achieved a 40%–45% major pathological response and 15%–16% pCR in resectable NSCLC.29 33 Contrarily, in the NADIM Study, patients receiving nivolumab combined with NACT showed a major pathological response of 83%, of whom 63% had a complete pathological response.31 Similarly, in breast cancer, a remarkably higher pCR rate for TNBC was observed in a series of studies on PD-1/PD-L1 inhibitors plus chemotherapy as NACT regimens, which increased from 22%–51.2% to 58%–64%.34–37 Collectively, these data highlight the therapeutic potential of PD-1/PD-L1 inhibitors in neoadjuvant therapy and provide the basis for the rational design of neoadjuvant chemo-immunotherapies for cervical cancer, even though no previous evidence is available. Selected ongoing trials of chemo-immunotherapies for LACC are summarised in online supplemental table 3.
The therapeutic activity of conventional chemotherapy is known to induce anti-cancer immunity by immunogenic cell death, which promotes the antigenicity and immunogenicity of tumours or by reprogramming tumour-reactive T cells and enhancing anti-tumour T cell responses. The standard therapies for cervical cancer include platinum-based drugs and taxanes, both known to have immunomodulatory effects. Cisplatin has been shown to upregulate MHC-I expression in cancer cells, thereby mediating antigen presenting to T cells. It can also remodel the immunosuppressive tumour microenvironment by depleting regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs).38 39 Meanwhile, cisplatin treatment upregulated PD‐L1 expression in both tumour and immune cells, thereby escaping from immune recognition and attack, but making an opportunity for the use of PD-1/PD-L1 inhibitors.40 In addition, docetaxel and paclitaxel have been shown to selectively reduce the number of MDSCs and Tregs without affecting the CD4+ and CD8+ T cell activity.41 Cisplatin plus paclitaxel therapy makes tumour cells more susceptible to cytotoxic T lymphocytes’ cytotoxic effect by increasing their permeability to granzyme B by upregulating of mannose-6-phosphate receptors on tumour cells.42 A similar conclusion was reached from clinical studies that cisplatin and paclitaxel treatment significantly decreased FoxP3+ Tregs and increased cytotoxic CD8+ T cells in the cervical tumour stroma,43 and NACT was associated with increased CD4, CD8, CD20 and CD56 signals, most prominently in good responders.44 Recently, we conducted a study to systematically assess the immune microenvironment before and after neoadjuvant chemotherapy, which showed that following the chemotherapeutic intervention, tumour-infiltrating immunostimulating cell concentrations were increased, including CD3+, CD4+ and CD8+ T cell, dendritic cells and macrophages. In contrast, the expression of the immunosuppressive factors FOXP3, IDO and PD-L1 was downregulated expression (figure 2). In general, chemotherapy reprogrammes the cervical tumour’s immunological microenvironment and makes the tumour more sensitive to immunotherapy.
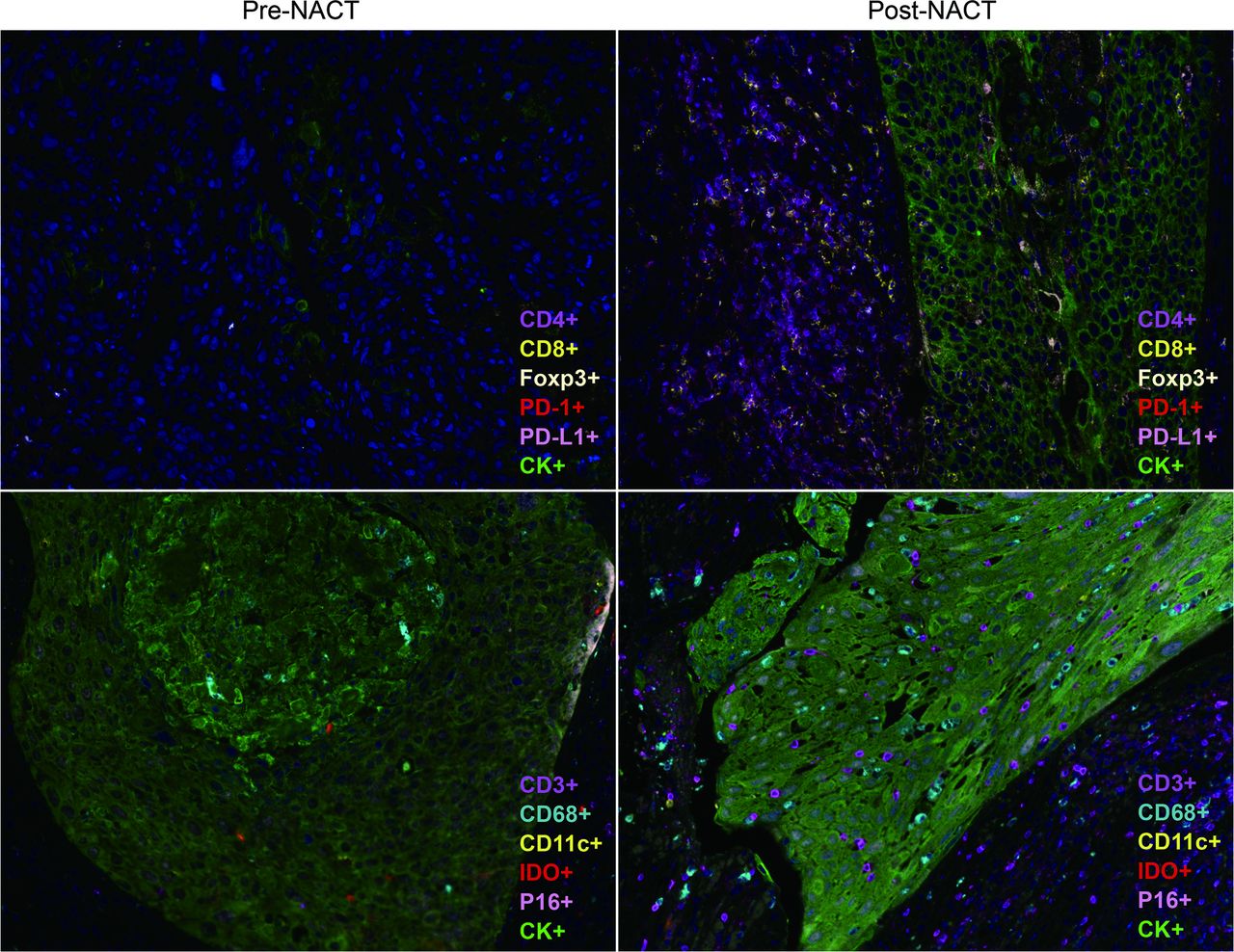
{kind=link}
{kind=link}
Representative image of a multiplex immunohistochemistry of the immune context of tumours of a patient with cervical cancer pre/post-neoadjuvant chemotherapy (NACT).
We conducted the first registered cohort study to evaluate the efficacy of neoadjuvant chemo-immunotherapy in LACC based on the promising clinical use and compact mechanisms of ICIs combined with chemotherapy. First, priming chemotherapy will be used to recondition the tumour’s immune microenvironment before anti-PD-1-combined chemotherapy. Next, PD-L1 detection is performed after enrolment and at the same time, patients receive platinum-based doublets of chemotherapy. Approximately 10 days later, the PD-L1 combined positive score result is used to determine whether they are enrolled in the clinical trial, without delaying the initial treatment of patients and increasing clinical feasibility. Third, paclitaxel is replaced with nab-paclitaxel as a chemotherapy regimen, avoiding the combined use of glucocorticoids that may affect the efficacy of PD-1 inhibitors, and can bring therapeutic benefits into fuller play.45 46 Besides, previous literature shows that two to three cycles of NACT could result in tumour shrinkage in 69.4%–79.3% of patients with LACC.7 20 Therefore, the one cycle of induction chemotherapy plus two courses of chemo-immunotherapy in our study are theoretically adequate to assess short-term efficacy without delaying the treatment for PD/SD patients. Finally, the postoperative treatments in this study are based on surgical pathological findings specified in the NCCN guidelines, rather than treating all patients with adjuvant therapy, as in other research,47–50 which is not only according to the international guidelines, but also avoids overtreatment suspicions and impact of different subsequent treatments on patient survival.
This study has some limitations. First, this is a single-arm study without a control group. Therefore, randomised controlled clinical trials involving large sample sizes and high quality should be conducted soon. According to ethical requirements, this study focuses on the PD-L1-positive subgroup, and the results should be generalised with great caution. However, broad literature has shown that PD-1 inhibitors are not limited to populations with positive PD-L1 expression,51 and subsequent studies will further expand the population.
In summary, the findings of this study will promote neoadjuvant anti-PD-1-combined chemo-immunotherapy with radical surgery as a new therapeutic strategy for patients with LACC. With the continuous improvement of medical care, treatment for cervical cancer is no longer monotherapy of radical surgery, chemotherapy or radiation. The integration of multiple treatments may achieve better cancer treatment effects. Further clinical trials and translational research are warranted to elucidate potential predictive biomarkers and investigate the mode of tumour microenvironment remodelling of anti-PD-1 therapies in cervical cancer. In this regard, patient stratification and individualised precision therapy can be achieved.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
JC and YH are joint first authors.
Contributors JC and YHan contributed equally to this trial and wrote the article. KL, GC and CS developed the study concept and protocol. YHu, XF, XM and SG assisted in further protocol development. KL supervises the clinical trial and has access to the final trial dataset. All authors contributed to the article and approved the submitted version.
Funding This trial is funded by the National Key Technology Research and Development Program of China (2022YFC2704400, 2022YFC2704403);the National Clinical Research Center of Obstetrics and Gynecology (2015BAI13B05),and Jiangsu Hengrui Medicine, Lianyungang, China (MA-CervC-II-002).
Disclaimer The funder had no involvement in the study design, the writing of this article or the decision to submit it for publication.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.