Article Text

Abstract
Introduction Surgical site infection (SSI) after abdominal surgery remains a significant cause of morbidity and is associated with an increased socioeconomic burden and a reduced quality of life. Circular wound protectors have been expected to reduce the risk of SSI, but previous studies reported conflicting results on their protective effects. The purpose of this study was to evaluate the efficacy of circular wound protectors in reducing SSI in open abdominal surgery.
Methods and analysis The circular pOlyethylen drape in preVEntion of suRgical site infection (COVER) trial investigates whether the application of a dual-ring circular plastic wound protector reduces the rate of SSI in patients undergoing elective or emergent open abdominal surgery related to the gastrointestinal tract, regardless of the type of wound classified by the Centers for Disease Control. The COVER trial is a multicentre, randomised controlled clinical trial with two parallel arms—one using a dual-ring wound protector with circular polyethylene drape and the other using conventional surgical dressing gauze. The primary outcome will measure the rate of SSI within 30 days after surgery in two groups. Statistical analysis of the primary end point will be based on the intention-to-treat population. The sample size was determined to achieve a study power of 80% with 95% two-sided confidence limits. Considering a dropout rate of up to 5%, a total of 458 patients, 229 patients in each group, will be enrolled in this study.
Ethics and dissemination The trial protocol and informed consent document have been reviewed and approved by the institutional review board at each participating centre. Written informed consent will be obtained from each study participant. The clinical outcomes of this trial will be submitted to an international peer-reviewed journal and presented at international conferences.
Trial registration number NCT 03170843.
- gastroenterology
- infection control
- surgery
- wound management
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This multicentre, randomised study includes elective and emergent surgery on the stomach, small and large intestine to ensure generalisability.
The primary endpoint, the 30-day postoperative surgical site infection (SSI) rate, will be assessed for open abdominal surgery with clean/clean-contaminated wounds and with contaminated/dirty wounds.
The Circular pOlyethylen drape in preVEntion of suRgical site infection trial will have the largest number of patients among all studies on a dual-ring, plastic wound protectors.
Wound condition will be assessed by the observer and by other investigators using wound photographs in the electronic case reporting form to provide reliable diagnosis of SSIs.
The limitations of this study are the lack of blinding of surgeons and the inclusion of only Korean individuals with a relatively low body mass index.
Introduction
Surgical site infection (SSI) is a common postoperative complication that is associated with considerable morbidity and mortality and with a significant socioeconomic burden.1–3 The rate of SSI is estimated to range from approximately 10% to 30% after elective abdominal surgery, depending on the presence of risk factors, type of procedure and degree of endogenous contaminants.1 4 5 In cases of faecal peritonitis, the SSI rate may reach up to 35~40%.6 7 Despite organisational, systematic approaches for preventing SSI based on evidence, such as preoperative antibiotic prophylaxis and antiseptic skin cleansing, SSI is still a major problem associated with increased hospital costs, prolonged hospital stays and unsatisfactory quality of life.8
The risk of developing an SSI will increase when the surgical incision site is exposed to large amounts of virulent bacteria in a contaminated surgical field.9 This risk has led to the idea of developing a physical barrier for the wound edge that can hinder direct exposure of the surgical incision edges to the contaminated field. Several devices that are designed for wound edge protection and have a similar design involving a flexible plastic wound cover placed in the laparotomy site are currently on the market. Prospective studies and randomised clinical trials (RCTs) have been conducted to evaluate the effectiveness of plastic wound protectors for reducing the incidence of SSI.8 10–13
Previous trials
The largest RCT evaluating the effectiveness of wound protectors in reducing SSI is the Reduction Of Surgical Site Infection using a Novel Intervention (ROSSINI) trial, with 760 patients undergoing laparotomy at 21 different hospitals in the UK.8 In this study, the drape type of wound protector was compared with standard intraoperative care. The results showed that the use of a wound edge protector during open abdominal surgery did not reduce the rate of SSI. Similarly, RCTs using a drape type of wound protector applied in colorectal surgery reported no benefit of the wound protector in reducing SSI.10 11 However, several other studies have claimed contrasting results. The BaFO trial, with 608 patients undergoing laparotomy at 16 different medical centres in Germany, demonstrated that the patients who used wound protection drape devices experienced SSI at a lower rate than those who did not.12 A Japanese single-centre RCT with 221 patients enrolled to investigate the effect of a double-ring, circular wound protector applied in non-traumatic gastrointestinal surgery also showed that the rate of SSI was significantly lower in the experimental group than in the control group.13
The effect of wound protectors in abdominal surgery is still controversial and remains to be elucidated. A well-designed, multicentre, RCT evaluating the effect of the dual-ring type of wound protector in open abdominal surgery, particularly for contaminated or dirty infected wounds, has not yet been conducted.
Methods and analysis
Objective
The COVER trial aims to investigate the effect of a dual-ring, plastic wound protector in open abdominal surgery. It is designed to test whether the device helps to reduce the overall rate of SSI development within 30 days postoperatively by 40% compared with that of the control group. In particular, the COVER trial includes patients who are undergoing an open abdominal surgery for contaminated or dirty/infected wounds, as well as those undergoing an open abdominal surgery for clean or clean-contaminated wounds, which allows a thorough investigation of the wound protector’s effects, depending on the degree of contamination.
Trial sites
Initially, eight sites at secondary or tertiary hospitals in South Korea began this trial. All participating investigators have been educated on the basis of the International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use, which serves as the good clinical practice (GCP) guidelines for this trial. This trial is still open for recruitment at participating centres.
Trial population and eligibility
All gastrointestinal surgical patients undergoing open abdominal surgery, either elective or emergent, will be screened for eligibility. Patients who satisfy the following criteria will be included: (1) between the ages of 18 and 75 years; (2) undergoing elective or emergent open abdominal surgery and (3) undergoing surgery on the stomach, small intestine or colon and rectum. Patients with any of the following will be excluded: (1) presence of concurrent infection in the abdominal wall; (2) open conversion from laparoscopic surgery; (3) presence of poor nutritional status indicated by a nutrition risk screening (NRS) 2002 score greater than 3; (4) undergoing combined hepatobiliopancreatic surgery; (5) pregnancy or breast feeding and (6) moderate to severe immunosuppression state, defined as previous organ or bone marrow transplantation, concurrent steroid administration (more than 10 mg prednisolone daily or an equivalent dose of any other steroid) or concurrent administration of other immunosuppressive or chemotherapeutic agents within the last 2 weeks prior to trial intervention. Once an investigator explains the extent and nature of the COVER trial to an eligible patient, informed consent will be obtained.
Trial type
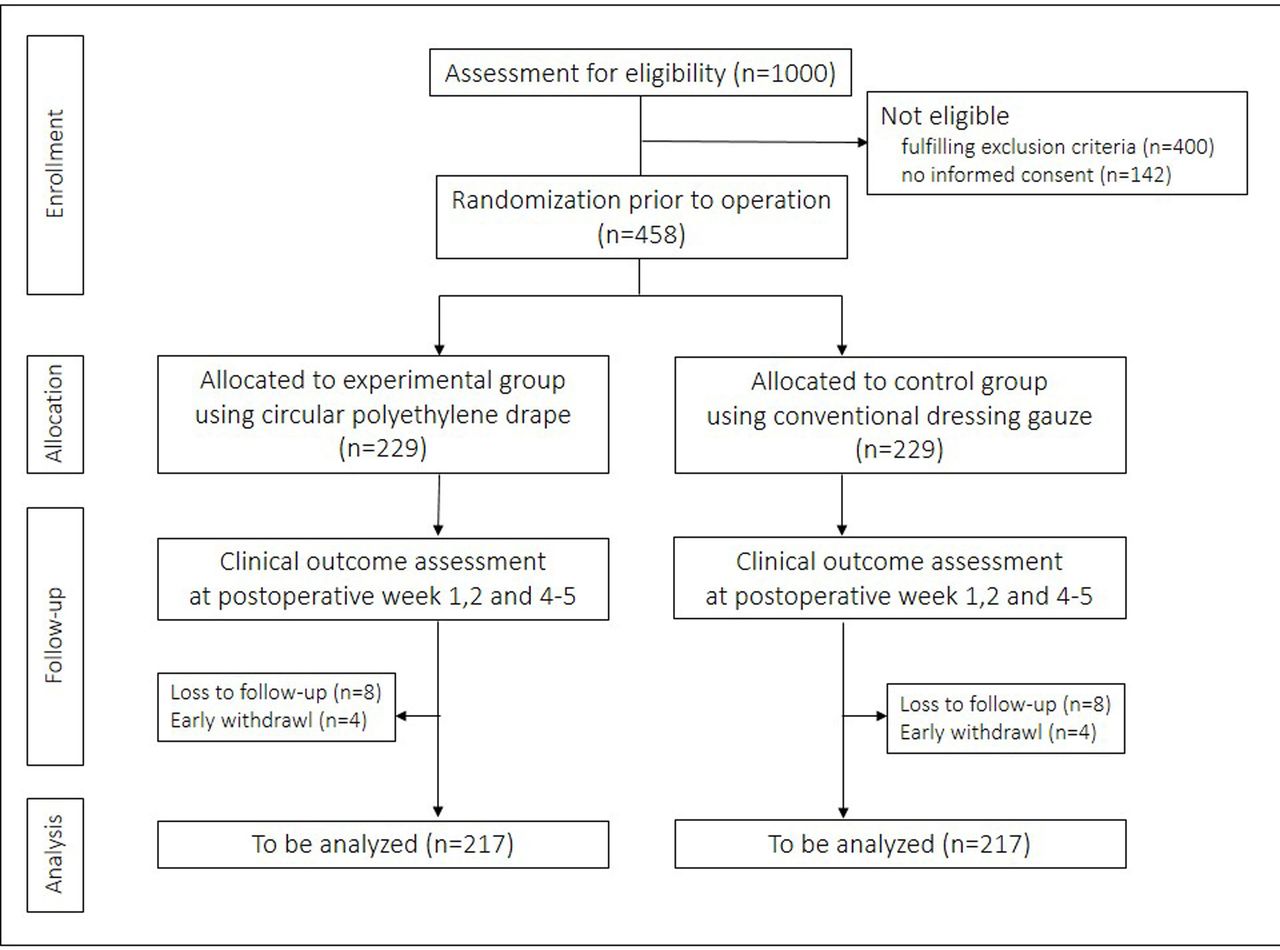
This clinical trial is a prospective, multicentre, patient-blinded, randomised controlled trial with two parallel comparison arms. A total of 458 patients will be enrolled, and 229 patients will be assigned to each group (figure 1).

Trial flow.
Recruitment and trial timeline
The eight centres at secondary or tertiary hospitals in South Korea have been actively conducting the trial since June 2017. Since then, four other centres have joined the trial recruitment, and this trial is still open for recruiting participating centres. All investigators, physicians or nurses are required to complete the ICH-GCP training course. Patients will be recruited for approximately 48 months. The last follow-up will be made 30 days after the last recruited patient undergoes the trial intervention. The SPIRIT figure shows the study schedule for enrolment, interventions and assessments (figure 2). A SPIRIT checklist is available in Additional file 1. An interim analysis is planned when 50% of the enrolment is reached. Depending on the results of the interim analysis, the subsequent research process and timeline can be modified.

{kind=link}
{kind=link}
SPIRIT figure. PO, postoperative; SSI, surgical site infection.
Randomisation and blinding
Stratification will be performed according to the participating centre and the type of wound classification. The wound types will be divided into two groups: one group with clean or clean-contaminated wounds and the other group with contaminated or dirty, infected wounds. A web-based patient registry (http://cover.e-trial.co.kr) will be applied to generate the allocation sequence immediately before the beginning of the operation, providing adequate concealment for the allocation sequence. The group allocation and randomisation number will be predefined by a biostatistician from the Catholic Medical Center in Seoul, South Korea. A permuted block randomisation with the size of two or four will be applied. Participating surgeons cannot be blinded to the allocated treatment. However, the patient will be blinded to the trial intervention since they are under general anaesthesia once the operation starts. The data manager will also be blinded because there is no direct access to either the trial intervention or the randomisation.
Interventions
Preoperative bowel preparation, type of skin preparation and drape, the use of perioperative antibiotics and the details of the surgical procedure will follow the policy of an individual surgeon at each centre. The experimental arm will be provided with a circular polyethylene drape (O Trac, Asung Medical, South Korea) to cover the incision site in the abdomen. It is a double-ring type of sterile, cylindrical wound protector consisting of inner and outer rings with a polyethylene sheath. The wound protector is left in situ throughout the operation and is removed immediately before closing the abdominal wall. The method of wound closure and insertion of wound drainage will also follow the policy of an individual surgeon at each centre.
For the control arm, conventional surgical dressing gauze will be used to protect the incision site during the surgical procedure. There are no differences in surgical technique, other devices or environment.
Risks
No additional risks to the participants are expected. The circular polyethylene wound protector has established clinical safety and has already been used in clinical applications with the approval of the Korean Medical Device Information and Technology Assistance Center. None of the technical details other than wound protection are affected by the trial.
Outcomes
The primary end point is the rate of SSI, defined by the diagnostic criteria suggested by the Centers for Disease Control (CDC), within 30 days after surgery. According to the CDC definition, SSIs are classified as either superficial incisional, deep incisional or organ/space.14 The postoperative wound condition will be evaluated at postoperative weeks 1, 2 and 4–5. The secondary end points include the length of postoperative hospital stay, the readmission rate and the rate of surgical complications other than SSI. The incidence of 30-day postoperative complications will be stratified according to the modified Clavien-Dindo classification.15
Data management and monitoring
A newly developed, web-based, electronic case reporting form (eCRF) will be used to record data for the included patients. Baseline characteristics, including age, sex, body mass index, American Society of Anesthesiologists score, history of smoking and alcohol consumption, history of previous chemotherapy, radiotherapy, abdominal surgery or steroid or immunosuppressive drug use, history of diabetes or malignancies in the gastrointestinal tract and nutritional status based on the NRS 2002 score, will be collected. Laboratory parameters (white cell counts and C-reactive protein and albumin levels) will be collected preoperatively, on the operation day and on postoperative day 2, if available. The parameters for the surgical procedure, including operation type (emergent or elective), site of operation (stomach, small intestine or large intestine), level of wound contamination according to CDC classification, method of skin preparation, antibiotic use, operation time, bowel anastomosis and stoma formation, wound closure material, length of skin incision, draining tube for the wound and body temperature during the surgical procedure, will be collected. The surgical wounds are classified as clean, clean-contaminated, contaminated and dirty wounds, according to the magnitude of the bacterial load.16 Postoperatively, the surgical wound will be evaluated at postoperative weeks 1, 2 and 4–5. A photograph of the wound will be taken at each office visit and documented in the eCRF. If SSI is detected, the classification and the postoperative date of diagnosis will be recorded. Bacterial culture of the infected wound will be performed. Postoperative complications according to the modified Clavien-Dindo classification, postoperative length of hospital stay and readmission will be noted. An investigator or research coordinator at each centre will enter the data using the eCRF. At the end of the trial, the study data and personal information of the enrolled patients will be archived for 3 years.
The trial data will be monitored by an independent institution (Medical Excellence) in Seoul, Korea. Monitoring will be performed in accordance with ICH-GCP guidelines.17
Safety evaluation and reporting of adverse events
All adverse events or serious adverse events, occurring from the moment of randomisation until the end of the 30-day follow-up, will be recorded and reported by the investigators.
Statistical methods
Sample size calculation
The sample size was calculated based on the primary end point of this trial. Previous reports on the incidence of SSI have indicated that the rate of SSI may vary depending on the wound classification, the procedure, the surveillance criteria and the quality of data collection.18 The incidence of SSI for clean/clean-contaminated wounds has been reported to be as high as 10%.19 For contaminated wounds, the incidence was approximately 25%.7 18 For dirty, infected wounds, the incidence may reach up to 40%.5–7 In this trial, the ratio of operations with clean/clean-contaminated, contaminated and dirty, infected wounds was assumed to be 20:40:40; therefore, the expected incidence of SSI for the control group was 28%. For the experimental group, the incidence of SSI will be decreased by 40%. Thus, the rate of SSI in the experimental group will be approximately 17%. The sample size was determined to achieve a study power of 80%, with 95% two-sided confidence limits. The actual sample size amounts to 434 participants. However, considering a dropout (lost to follow-up, retracted consent or protocol violation) rate of up to 5%, a total of 458 patients, 229 patients in each group, will be enrolled in this study.
Statistical analysis
The statistical analysis will be performed by an independent statistician from the Catholic Medical Center (Seoul, South Korea). The interim and final results will be analysed mainly for the intention-to-treat population and, additionally, for the per-protocol population. The rate of 30-day postoperative SSI will be evaluated in all patients and analysed according to the wound classification (superficial incisional, deep incisional and organ/space SSIs), as defined by the CDC. Pearson’s χ2 test or Fisher’s exact test will be used to analyse nominal data; Student’s t-test and the Wilcoxon rank-sum test will be used for continuous data. To estimate the independent risk factors for 30-day postoperative SSI, logistic regression analysis will be performed. The statistical analysis will be conducted using SAS V.9.4 (SAS Institute).
Withdrawals
Enrolled patients can withdraw their participation at any time, if desired. In this case, the patients will have no disadvantages. The investigator will record any patient withdrawal in the eCRF.
Patient and public involvement
Patients and the public were not involved in the protocol of this study.
Ethics and dissemination
Research ethics
Written informed consent was obtained from each study participant in accordance with ethical approval.
Dissemination
The final results will be discussed with participating surgeons and presented at domestic and international scientific conferences. The final results will be submitted to an international peer-reviewed scientific journal.
Discussion
SSI has been recognised worldwide as a costly, debilitating surgical complication for decades. Despite vigorous efforts to control SSI through campaigns and publications by international organisations, the rate of SSI has changed only slightly.2 20–23 Even such recommendations are limited to the use of prophylactic antibiotics or antiseptic skin cleansing, which can be applied only during elective surgeries. In cases of abdominal surgery, diffuse purulent peritonitis with or without faecal contamination, which requires emergency surgery, is frequently encountered. Prophylactic antibiotics or antiseptic skin cleansing is not applicable in emergent surgical cases. Several preventive measures other than the use of prophylactic antibiotics or antiseptic skin cleansing have been proposed to prevent SSI. Intraoperative wound irrigation with antibiotic solution is one method that can be implemented. Intraoperative wound irrigation with antibiotic solution seems to reduce the incidence of SSI; however, there are potential adverse effects of tissue toxicity and increased bacterial resistance.24 Another method is the application of negative-pressure wound therapy (NPWT) without primary closure of the abdominal wound in highly contaminated abdominal surgery.25 A recent meta-analysis on the use of NPWT in open and infected wounds after vascular surgery demonstrated that it could be effective in controlling SSI.26 However, there are only a few case reports on its use in contaminated abdominal surgery, and no trial or analysis of its efficacy is available. The first two methods require the application of a bactericidal substance directly to the tissue that may or may not present a bacterial infection. Thus, the adverse effects of tissue toxicity and bacterial resistance cannot be ignored. The use of NPWT also requires additional resources and time to heal, which potentially involves a longer hospital stay and additional medical costs. Therefore, adopting these methods is not easy in daily practice.
The application of a plastic wound protector in abdominal surgery has been tested for its efficacy for more than a decade. Based on findings for the pathogens most frequently isolated in SSI, including Staphylococcus aureus, coagulase-negative staphylococci, Enterococcus species and Escherichia coli,18 plastic wound protectors that hinder direct exposure of the surgical wound to virulent endogenous bacteria during surgical procedures have been created. Several previous studies and trials have been conducted to investigate this hypothesis.27 These trials have varied by using different designs of wound protectors: namely, single-ring or dual-ring types. A meta-analysis by Mihaljevic et al showed that wound edge protectors significantly reduced the rate of SSIs in open abdominal surgery, but the available data for double-ring wound protectors might be lower quality than those available for the single-ring device.28 The COVER trial will test a dual-ring type of wound protector that can tightly conceal the surgical incision edge during the entire operation time. Previously, the trials on the dual-ring design were conducted in a single centre with a small sample size. In addition, these trials excluded emergent surgeries with contaminated and dirty, infected wounds resulting from perforated viscera.13 29 30 Therefore, the effectiveness of the dual-ring type of wound protector in controlling SSI in contaminated and dirty, infected wounds can be addressed. In the COVER trial, patients more than 75 years will be excluded. Prevalence of cognitive impairment increased with age in patients more than 75 years,31 and these patients often have difficulties in understanding the concepts of clinical trial. In addition, extreme age itself is associated with an increased risk of SSI.32
The COVER trial is a pragmatic, two-armed RCT that will be conducted by at least 11 surgeons at 11 different centres and possibly more, which will increase external validity. Internal validation and data quality will be ensured by adherence to the SPIRIT statement.33 Assessments of wound condition will be performed by the observer and reviewed by other investigators via photographs documented in the eCRF. This will provide an objective and reliable method for the evaluation of wound infections.34 Finally, the risk that patients may experience from participating in this trial is minimal and will remain within the boundaries of routine clinical practice.
The results of the COVER trial will provide high-quality evidence for the use of a circular polyethylene drape in open abdominal surgery with all types of wounds to reduce the incidence of SSI.
Trial status
Recruitment of participants began on 11 July 2017. A total of 211 patients were recruited for this trial as of 21 September 2019. The trial is currently ongoing. (current study protocol V.7.0., revised on 23 October 2018)
References
Footnotes
Contributors HJK and RNY designed the COVER trial and were responsible for the protocol development.JIL, W-KK, B-HK, CWK, SUB, SMN and BMK revised the draft of the protocol and approved the final version of protocol. RNY and BMK wrote the manuscript. BMK and HJK critically revised the manuscript.All of the authors conducted the COVER trial and approved the final version of manuscript.
Funding This trial is supported by the Korean Surgical Infection Society (Award number: KSIS 2019-021), with the use of the circular polyethylene wound protector (O Trac, Asung Medical Inc. South Korea) given free of charge. There is no other financial support or conflict of interest. The industrial funder and trial management are independent.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.