Article Text

Abstract
Introduction Chronic non-malignant pain has a major impact on the well-being, mood and productivity of those affected. Opioids are increasingly prescribed to manage this type of pain, but with a risk of other disabling symptoms, when their effectiveness has been questioned. This trial is designed to implement and evaluate a patient-centred intervention targeting withdrawal of strong opioids in people with chronic pain.
Methods and analysis A pragmatic, multicentre, randomised controlled trial will assess the clinical and cost-effectiveness of a group-based multicomponent intervention combined with individualised clinical facilitator led support for the management of chronic non-malignant pain against the control intervention (self-help booklet and relaxation compact disc). An embedded process evaluation will examine fidelity of delivery and investigate experiences of the intervention. The two primary outcomes are activities of daily living (measured by Patient-Reported Outcomes Measurement Information System Pain Interference Short Form (8A)) and opioid use. The secondary outcomes are pain severity, quality of life, sleep quality, self-efficacy, adverse events and National Health Service (NHS) healthcare resource use. Participants are followed up at 4, 8 and 12 months, with a primary endpoint of 12 months. Between-group differences will indicate effectiveness; we are looking for a difference of 3.5 points on our pain interference outcome (scale 40 to 77). We will undertake an NHS perspective cost-effectiveness analysis using quality adjusted life years.
Ethics and dissemination Full approval was given by Yorkshire & The Humber - South Yorkshire Research Ethics Committee on 13 September, 2016 (16/YH/0325). Appropriate local approvals were sought for each area in which recruitment was undertaken. The current protocol version is 1.6 date 19 December 2018. Publication of results in peer- reviewed journals will inform the scientific and clinical community. We will disseminate results to patient participants and study facilitators in a study newsletter as well as a lay summary of results on the study website.
Trial registration number ISRCTN49470934; Pre-results.
- chronic non-malignant pain
- opioids
- self-management
- behavioural interventions
- tapering
- RCT
- process evaluation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- chronic non-malignant pain
- opioids
- self-management
- behavioural interventions
- tapering
- RCT
- process evaluation
Strengths and limitations of this study
First large multicentre randomised controlled trial in UK to test an active intervention (group based multicomponent self-management programme plus one-to-one support) in comparison to control (self-help booklet and relaxation compact disc) in opioid tapering for those with chronic non-malignant pain.
Recruitment is mainly through primary care, where a large population can be screened for use of strong opioids.
The intervention is manualised, comprehensive and includes a specifically designed app used by the clinical facilitators to generate opioid tapering plans.
The embedded process evaluation will help us understand people’s experiences of the intervention and what helped or hindered its use (compared to the control group).
The proposed best usual care method is not embedded in current National Health Service practice and may thus not represent usual care model.
Introduction
Chronic pain is defined as pain that persists past normal healing time of around 12 weeks1 2 and affects eight million people (15%) in England alone.3 Around 20% of those aged 34 years old or over, and around 40% in those aged 75 years old or over, report high levels of interference with their lives from pain.3 The common disorders contributing to this include low back pain, neck pain, osteoarthritis, neuropathic pain, fibromyalgia, chronic widespread pain and postsurgical pain. Individuals may live with more than one of these pain disorders. While opioids are regularly prescribed for the management of chronic non-malignant pain, they are not always effective in the long term and can cause a range of adverse effects such as sedation, nausea, respiratory depression/sleep apnoea, depression, abdominal pain, overdose and even death.4 5 Furthermore, people on long-term opioid treatment (3 months or more) report inadequate analgesia, in spite of high doses, due to the development of tolerance with reduced function, quality of life or absence of progress toward therapeutic goals.6–8 Yet, prescription data from the UK show that over an 11 year period (2002 to 2013) there was an increase in prescribing of the more potent controlled and long-acting, long-term opioids for those with musculoskeletal conditions within the first 90 days of their long-term episode (2.3% to 9.9%).9 There is a pressing need for interventions to help people withdraw from strong opioids used for chronic non-malignant pain.
While much is known about the adverse effects of long-term opioid treatment,10 little is known about the economical impact of these adverse events. There are few evaluations of interventions designed to support opioid reduction. Cochrane reviews11 12 and randomised controlled trials13 14 offer some support for interventions supporting opioid withdrawal, including interdisciplinary pain management programmes, use of behavioural strategies, motivational interviewing, mindfulness and pain education, however, this is of low quality with short follow-up (≤4 months).15
There are no formal UK guidelines for opioid reduction in this population. While such recommendations are currently emerging in North America,16 17 these are based on expert consensus rather than data. There is also no clear evidence to support a particular speed of opioid tapering or the use of particular opioid drug(s) or rotating from one opioid to another. Overall, data substantiating the role of self-management and cognitive behavioural interventions in support of opioid tapering is weak and mostly applicable to the North American health systems.17 Consequently, this trial will test an evidence-based intervention for people with chronic non-malignant pain. The intervention is designed to help patients manage pain interference, reduce individuals’ opioid consumption and enhance quality of life.
Methods/design
Trial design and objectives
The primary objective is to test the clinical and cost-effectiveness of a patient-centred, group-based, multicomponent, self-management intervention for people living with chronic non-malignant pain, compared with a best usual care (ie, the control group intervention) in a two-arm pragmatic randomised controlled trial (figure 1). The intervention targets withdrawal of strong opioids to assess the impact of their withdrawal on pain interference with daily living. We are running an embedded process evaluation (publication in preparation) to test fidelity to inform the interpretation of the findings and, if indicated, implications for the implementation of the intervention across the National Health Service (NHS).
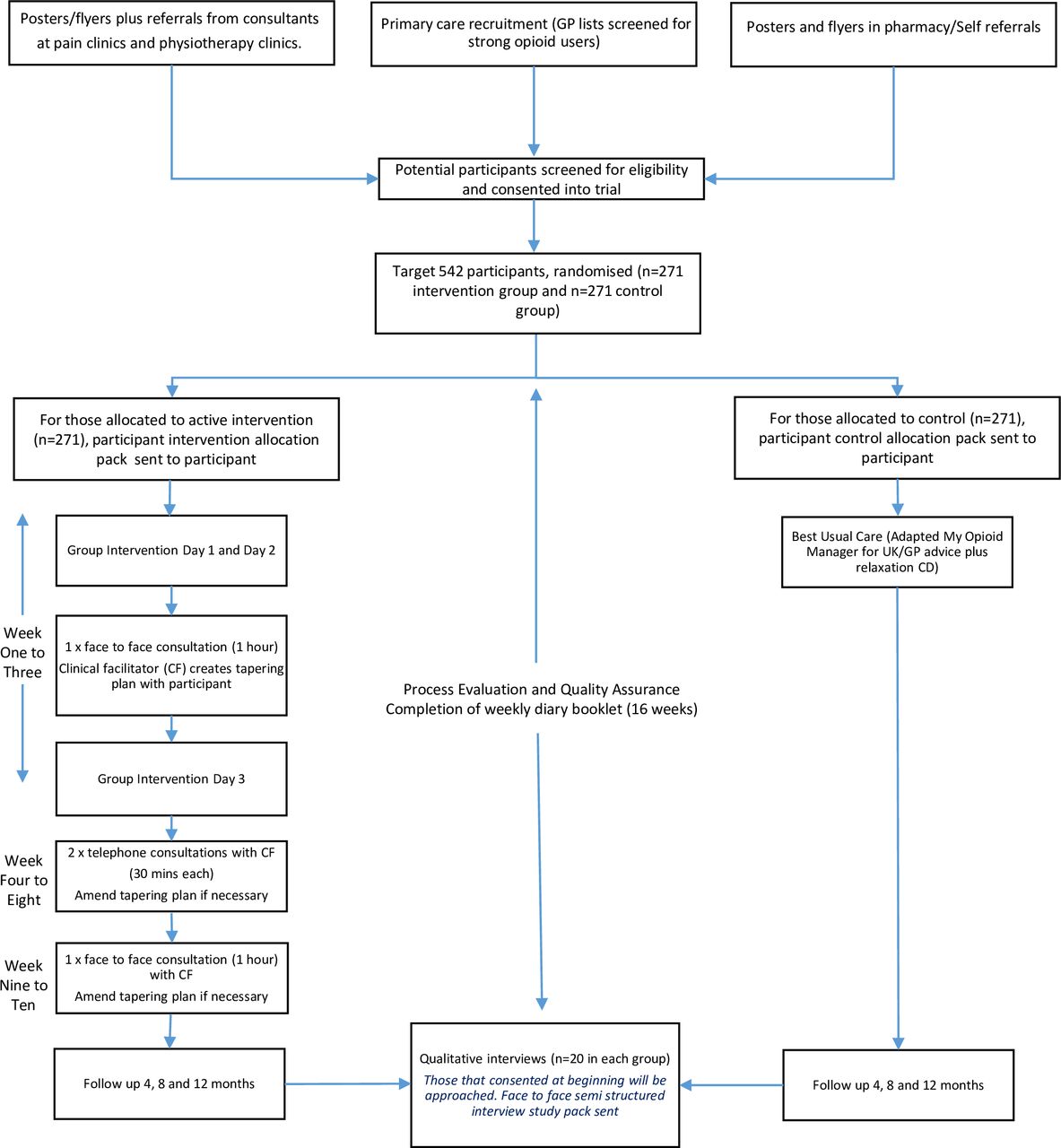
{kind=link}
I-WOTCH flow chart. GP, general practitioner.
Dates of study enrolment are: 17 May 2017 to 30 January 2019.
Trial setting
The trial is taking place in the North East, East Midlands, West Midlands and South Central areas of England. When originally planned we also intended to recruit in London. However, operational barriers meant we were unable to deliver the trial in London. The populations from which participants are drawn are broadly representative of the UK. We are recruiting participants from general practices, community pain services, local musculoskeletal services and pharmacies. We also accept self-referrals (eligibility is checked over the phone once an expression of interest form is returned to check study criteria are met including non-cancer pain diagnosis and use of strong opioids). All medications reported on the baseline questionnaire are checked again with the participant at time to consent. Recruitment sites are clustered by reasonable geographical proximity to a treatment site and people in one locality are approached accordingly so that the intervention groups can be populated in a timely manner.
For the purpose of this study, we define strong opioids using the British National Formulary (BNF). Thus, we are recruiting participants taking any of the following analgesia: buprenorphine, dipipanone, morphine, diamorphine, fentanyl, hydromorphone, methadone, oxycodone, papaveretum, pentazocine, pethidine, tapentadol or tramadol.18 Although in some jurisdictions tramadol is considered to be a weak opioid the BNF classifies it as a strong opioid.
Recruitment procedures
(Primary care) electronic screening of general practitioner (GP) records: GP practice lists are searched electronically to identify people (aged 18 years or over), who have been prescribed strong opioids on more than one occasion in the previous 3 to 6 months and in the previous 0 to 3 months as indicated by their health record and not in a care home or housebound. The practice then screens the list of those identified from the first search to exclude patients taking strong opioids for malignant pain or who should not be approached for other reasons, including those at risk, vulnerable or not suitable for a group-based intervention. Those who are using methadone for purposes other than to manage chronic pain are not approached.
Referred to the study by their GP or healthcare professionals at pain clinics and musculoskeletal physiotherapy clinics. GPs and healthcare professionals at pain clinics and musculoskeletal physiotherapy clinics can also refer potential participants by giving them an information pack on the study and an expression of interest form.
Posters advertising details of the study are displayed in prominent areas of GP surgeries, pharmacies, pain clinics and musculoskeletal physiotherapy clinics. The posters contain information about the study, including contact details for the study team.
Eligibility and informed consent
Once we receive an expression of interest form from a potential participant (with contact details), a member of the study team contacts them by telephone to check their eligibility for the study, using the inclusion and exclusion criteria as a checklist (table 1). Pregnancy or actively trying for pregnancy was included as an exclusion from 15 November 2018. Following a query from a potential participant we identified some evidence, from the addiction literature that abrupt opioid withdrawal may trigger miscarriages and stillbirths.19 20 There is no clear evidence on how to taper opioids in pregnancy and detoxification is not recommended as a treatment intervention due to limited evidence available, low completion rates of detoxification and high rates of relapse.21
Eligibility criteria
During this telephone check the study team give a brief background as to why the study is being done and specifically the aims of opioid reduction and study design. At this stage of enrolment commitment to opioid dose reduction is not mandatory, the aim is to give potential participants as much information as possible and answer any questions they may have. An anonymised screening log is kept, detailing all those screened and reasons for exclusion. Potential participants (those who are eligible) are then sent a study pack in the post containing an I-WOTCH cover letter, participant information sheet, trial consent form (online supplementary file), baseline questionnaire and pre-addressed envelope. On receipt of a signed consent form and completed questionnaire, a designated member of the study team performs a final telephone eligibility check on medications reported by the patient in the baseline questionnaire. If the medications meet the eligibility criteria and consent is deemed to be valid and informed, the consent form is countersigned by the appropriately trained member of the study team and a copy of the completed form is sent to the participant and to the participant’s GP. Participants are informed that if they decline to take part in the study or are found to be ineligible there is no impact on their usual form of care or access to opioid medication. Where possible, participants are randomised in batches of 24 to ensure adequate group sizes. Once they are randomised they are sent a letter informing them of this with details of the group session (time and venue) if they are randomised into the intervention arm. There is no further checking or consent at this point.
Supplementary material 1
Experimental intervention
I-WOTCH is an 8 to 10 week programme with a mixture of group sessions (facilitated by a trained I-WOTCH clinical facilitator, usually a nurse, and a trained lay person with chronic pain and experience of opioid tapering, or an I-WOTCH trained allied healthcare professional), two one-to-one consultations and two telephone calls with the I-WOTCH trained clinical facilitator. The programme is adapted from a previously tested intervention used for the self-management of chronic pain.22 23 All those randomised to the I-WOTCH intervention in a locality are invited to join Day 1 of the next available programme. Should no group be available, or if a participant withdraws from the group based intervention, they are sent all of the written material that they would have received had they attended the group. Attendance at Day 1 is mandatory for accessing subsequent elements of the programme.
One-to-one clinical facilitator consultations
Between Day 2 and Day 3 of the group based sessions, participants attend a face-to-face, consultation with the I-WOTCH trained clinical facilitator. This is an opportunity for the clinical facilitator to explore opioid tapering experiences with participants, including thoughts, motivation, perceived challenges and opportunities, and to collaboratively develop realistic tapering goals adapted to the participant’s circumstances. The clinical facilitators are trained to use motivational interviewing skills to facilitate discussion. After Day 3 participants receive two telephone consultations (approximately 30 min each) to discuss progress with the tapering and to identify the need for other support during withdrawal. They also receive a final face-to to-face consultation to (i) reflect on progress, (ii) recap over self-management skills covered in the group sessions, (iii) review and reset goals and objectives and (iv) assess future needs for support.
I-WOTCH tapering app to generate the opioid tapering plan
We have developed a tapering app to be used by the I-WOTCH clinical facilitators in the one-to-one consultations to generate the tapering plan. The app was developed within the programming team at Warwick Clinical Trials Unit with clinical expertise guided by SE. The app facilitates calculations of tapering regimes, as well equianalgesic doses of systemic opioids when switching between opioid preparations is necessary. During our preparation for the study and design of the app we uncovered a discrepancy between a number of existing equianalgesic opioid tables24. For the purposes of opioid tapering we used the Faculty of Pain Medicine equianalgesic table (https://www.rcoa.ac.uk/faculty-of-pain-medicine/opioids-aware/structured-approach-to-prescribing/dose-equivalents-and-changing-opioids) for our calculation.25 We supplemented this with reviews of the individual drugs’ summary of product characteristics and other sources where needed.26 27 For the purposes of managing changes in medication during the taper, individual variability is taken into account. Once the tapering plan is generated a paper copy is given to the participant and the electronic data is sent to the study team for checking and filing. All tapering plans generated are checked by a clinician for accuracy (SE or JN). A paper copy is sent to the participant’s GP for their records.
Opioid tapering procedures
Participants are tapered on their drug of presentation. Opioid rotation is only recommended for participants who have reached the lowest dose of a transdermal patch preparation. For example, in cases of participants presenting on fentanyl transdermal patches these are tapered in decrement of 12 mcg/hour patches, and an oral formulation of alternative opioid with equianalgesic potency introduced when the lowest increment of the patch is reached.28 Participants on buprenorphine patches are weaned in decrements of the patches with no substitution due to its agonist/antagonist action.29 The nurses make every effort to encourage participants to stick to the suggested tapering schedule. Nevertheless, there is an element of negotiation about speed, if any, of dose reduction.
We are using a regimen based on the Mayo Clinic experience as it provides some evidence to support the notion of slow tapering and is unlikely to be associated with severe withdrawal symptoms and therefore likely to facilitate adherence.28 This consists of a 10% decrease of the original dose every 5 to 7 days until 30% of the original dose is reached. This is followed by a weekly decrease by 10% of the remaining dose. The 10% may be rounded up or down to suit prescribing. We are providing training in equianalgesic dose calculation as well as an electronic means of calculating. People utilising opioids as rescue analgesia, at a frequency of less than one dose per day, do not require a formal tapering regime but are still being supported to completely withdraw from opioids.
Facilitator training
I-WOTCH facilitators (clinical and lay) attend a 2-day training course delivered by HS and JS (experienced in the design and delivery of the intervention) to deliver the intervention. Over the 2 days the facilitators are taught how to deliver each of the topics, as well as given an opportunity to experience the mindfulness and relaxation practice which is a part of the programme. In addition they are also taught group facilitation skills and procedures to follow within the study. The I-WOTCH clinical facilitators also attend a third day of training, during which they are given further opioid education, and trained in how to taper, use of the I-WOTCH app, motivational interviewing and study procedures for the one-to-one consultations.
We have adapted a comprehensive facilitator’s manual and training programme used in a previous trial on the management of chronic pain23 to facilitate delivery of the I-WOTCH intervention (in preparation). The adaptations have been formed through literature, piloting of the intervention and input from lay people (those with chronic pain and experience of opioid use). The manual is acting as a guide and a reference point for all facilitators throughout the intervention.
Adaptations and development of the intervention has included: structuring the programme to include opioid education as well as pain education, and integrating these throughout the programme. Specific examples and case studies related to opioid tapering and pain have been used, mindfulness and relaxation compact discs (CDs) created and a video (focused on pain and opioid education) has been produced for participants. Clips of the video are integrated into the group programme to illustrate specific topics such as pain education, challenging unhelpful thoughts and fear related to opioid tapering and withdrawal. Participants are given the full video on a digital versatile disc (DVD) to watch with their friends and family. Having the DVD allows the participant to watch it in their own time and consolidate the information learnt on the programme. Specially designed handouts are given to participants at the end of each group day summarising key topics discussed. Further details of the intervention and its theoretical framework used to design are to be reported elsewhere.
Control intervention
Those randomised to the control group will receive augmented usual care, including two participant-facing components: a hard copy of the I-WOTCH adapted ‘My Opioid Manager’ booklet, and a relaxation CD with instructions on its use. ‘My Opioid Manager’ was developed in Canada (Toronto Rehabilitation Institute) specifically for people using opioid drugs for chronic non-cancer pain. It is a self-help guide that contains information about opioids and provides guidance about setting goals, issues the participant may encounter, tapering and non-opioid options for management of chronic pain. It is based on the 2010 Canadian Opioid guideline of opioids for chronic non-cancer pain.30
Compliance
For the intervention, we are recording the number of sessions that each participant attends, including the follow-up calls completed. Assurance checks through the study also include the integrity of randomisation, study entry procedures and data collection.
Study outcomes
Primary outcome: activities of daily living
Our primary clinical outcome is the Patient-Reported Outcomes Measurement Information System (PROMIS) Pain Interference Short Form (8A) (PROMIS-PI-SF-8A).31 This is an eight-item generic self-reported measure, which assesses the consequences of pain on relevant aspects of an individual’s life and key activities of daily living: engagement with social, cognitive, emotional, physical and recreational practices. The PROMIS-PI-SF-8A raw score ranges from 4 to 80 which is then standardised to give a score ranging from 40.7 to 77, with higher scores indicating worse outcome. The PROMIS-PI measures the same construct as two legacy pain interference measures (Brief Pain Inventory Pain Interference subscale and the SF-36 Bodily Pain subscale), supporting the calculation of a common metric.32 33
As originally designed, we proposed pain interference as a single primary outcome to ensure we could recruit sufficient participants. However, how the intervention might affect pain interference directly through its behavioural and educational component and how there might be indirect effects through changing opioid use is unclear. It is possible, for example, that the intervention has a good effect on opioid use but little effect on pain interference. In this situation the potential long-term benefits of opioid reduction might justify claiming a positive result from the trial. During the later stages of recruitment, it became clear we had capacity, and sufficient interest from practices and potential participants to exceed our planned target. This allowed us to add opioid use as a prespecified second primary outcome.
Our main outcome measure for opioid use is the mean difference in morphine equivalent dose in the 4 weeks prior to 1 year follow-up expressed as mg equivalents of morphine per day. In ongoing work we are reviewing morphine equivalence tables before making a final decision on which set of equivalence values to use for this analysis. For sensitivity analyses, we will use alternative published values for equianalgesic doses of opioids to ensure that our findings are robust if different weightings are used. For secondary analyses, we are comparing proportions achieving a complete withdrawal and proportions of responders, defined as ≥50% reduction in morphine equivalent doses taken, between intervention and control groups.
While our study entry criterion is participant reported use of strong opioids on most days in the preceding 4 weeks, our continuous measure of opioid use is mean morphine equivalents of opioid used in the preceding 4 weeks. This includes all opioids consumed, including any weak or as required opioids.
Self-reported data on opioid use are being collected at baseline, 4, 8 and 12 months following randomisation via postal follow-up. At baseline, one postal reminder is sent. At 4, 8 and 12 months a postal reminder is sent. In the event that no response is obtained from the postal reminder at 4, 8 or 12 months, we contact the participant by phone and collect our primary clinical outcome, opioid use and EQ-5D-5L34 over the phone. Participants complete a weekly diary that includes the EQ-5D-5L34 and the Short Opiate Withdrawal Scale35 for the first 4 months after randomisation.
Secondary outcomes
Our package of other secondary outcomes and process measures is informed by the consensus recommendations for core outcome domains for trials of the efficacy and effectiveness of treatments for chronic pain by the Initiative on Methods, Measurement and Pain Assessment in Clinical Trials group.36 All outcome measures are presented in table 2 with data collection time points.
Outcomes measures and time points
Power and sample size
For the purposes of our original sample size calculation we used our primary clinical outcome measure, the PROMIS-PI-SF-8A.31 Using the PROMIS primary outcome, participants in the control arm are likely to obtain a mean score of 50, SD 10.37 A sample size of 346 participants is required to show a difference of 3.5 points on PROMIS-PI-SF-8A (standardised mean difference of 0.35) at 5% significance with 90% power. There may, however, be clustering effects by groups in the intervention arm. We do not have any data from similar studies to inform an estimate of the intracluster correlation (ICC). Our recent experience across multiple studies of group interventions has been that such effects are trivial or negligible.22 38 39 Despite this, assuming a relatively modest ICC of 0.01 and assuming, on average, that 10 participants per group provide 1 year outcome data, we would require 374 patients. Allowing for 20% loss to follow-up (while striving for 10%) we planned to recruit 468 participants. We subsequently changed the significance level to 2.5% to allow for two primary outcomes (PROMIS-PI-SF-8A and opioid use) with effect size of similar magnitude, and adjusted the inflation factor for clustering to reflect actual group sizes. Our final recruitment target was 542 participants. Experience in similar studies is that, towards the end of recruitment, there can be a need to over-recruit slightly more people than originally projected to ensure the final intervention groups are adequately populated.
Randomisation methods and blinding procedures
Patients are randomised in a 1:1 ratio to either the I-WOTCH intervention or best usual care arms. Randomisation procedures are being performed at the Warwick Clinical Trials Unit (WCTU) where possible randomisations are carried out by a member of staff who is not a core member of the I-WOTCH team. The method of randomisation is computer generated using WCTU randomisation systems developed by the WCTU programmers. There is no allocation concealment as the person conducting the randomisation is also entering baseline data prior to randomisation. All baseline data are being collected prior to randomisation. Where possible any data collected from GP records is being done by staff blind to treatment allocation. Routine data sources such as GP prescribing data are also collected.
To ensure that we populate the groups, we are clustering groups of four to five geographically proximate practices with approximately 50 000 patients to launch recruitment at around the same time. We are then randomising participants when we have sufficient participants to populate a group in batches of around 24 participants to minimise time between randomisation and the start of the intervention. We aim to randomise within 2 weeks of the start of the group. This ensures exposure time to the two interventions is as similar as possible. We anticipate the number of people in a group to between 10 and 12 participants. However due to the pragmatic nature of the trial and the possibility that participants may not turn up on the day of the group, we will run the group with a minimum of two people. We will record attendance and the number of people in each group for each day. Randomisation has been stratified by group (Groups 1 to 35), baseline pain severity (PROMIS 3A score: low pain=3 to 8, high pain=9 to 15) and baseline opioid use (morphine equivalent dose: 0 to 29, 30 to 59, 60 to 89, 90 to 119, 120 to 149, 150+).
Data management
Data for individual participants are being collected via participant-completed questionnaires, by clinical facilitator-completed case report forms (CRF), or by collection from participants’ GP records by a member of the I-WOTCH research team or local clinical research network support team.
Participant identification in the CRF is done through their initials and unique research number allocated at the point of entering into the study. Data are being collected from the time the potential participant is considered for entry into the research through to completion of the intervention and follow-up period (interviews are conducted after the 12 month follow-up). Data are subject to a full set of validation checks and additional data checking procedures to assure quality of data entry.
All (paper) data are being held securely by the research team at WCTU for the baseline questionnaires, intervention evaluation sheets, postal questionnaires at 4, 8 and 12 months, weekly diary booklets and any ad hoc CRFs required. The database has been developed by the programming team at WCTU and all specifications (ie, database variables, validation checks, screens) have been agreed between the programmer and appropriate trial staff including the trial statistician.
All essential documentation and trial records are being stored by WCTU in conformance with the applicable regulatory requirements. Access to stored information is restricted to authorised personnel only.
We developed questionnaires to record relevant information. CRFs have been designed by research fellows and the trial manager in conjunction with our trial management group (TMG) building on the expertise of the applicants. The TMG consists of project staff and co-investigators involved in the running of the day-to-day trial. Significant issues arising from management meetings are referred to the Group.
The trial steering committee (TSC) has an independent chairperson. The Committee is responsible for major decisions such as a need to change the protocol for any reason, monitoring and supervising the progress of the trial, reviewing relevant information from other sources, considering recommendations from the Committee and informing and advising on all aspects of the trial.
The data monitoring committee (DMC) consists of independent experts with relevant clinical research and statistical experience. Confidential reports containing recruitment, protocol compliance, safety data and interim assessments of outcomes are being reviewed by the Committee. The DMC is responsible for monitoring data and making recommendations to the TSC on whether there are any ethical or safety reasons why the trial should be amended or terminated. The DMC will determine if additional interim analyses of trial data should be undertaken, and if so, when. The DMC will meet early on in the trial and then annually or more frequently if necessary. The final DMC meeting will be held on the availability of the final trial data.
Both the trial steering committee and the data monitoring committee follow WCTU standard operating procedures.
All electronic participant-identifiable information are held on a secure, password-protected database accessible only to essential personnel. Paper forms with participant-identifiable information are held in secure, locked filing cabinets within a restricted area of WCTU. Participants are identified by a unique research number only. Direct access to source data/documents is required for trial-related monitoring. For quality assurance, the data and results are statistically checked. A full data management plan has been produced by the trial manager and statistician to outline the data monitoring checks required. Trial documentation and data will be archived for at least 10 years after completion of the trial.
Adverse event management
Any adverse events are reported to the trial coordinating centre by the clinical facilitators in each region within 24 hours of them becoming aware of the event. Participants will be asked if they have experienced any adverse effects while tapering opioid use at the clinical facilitator consultations, in the weekly diaries and in questionnaire follow-up at 4, 8 and 12 months; and if so, which symptoms they have experienced. Participants GP’s will not be informed of any adverse events unless there are serious safety concerns and there is a chance of significant harm to the participant or others. In accordance with WCTU standard operating procedures risk assessment is completed and a trial monitoring plan produced commensurate to the risks identified.
Statistical analysis
The data will be summarised and reported in accordance with Consolidated Standards of Reporting Trials guidelines for randomised controlled trials. We are using intention-to-treat analyses.40 Hierarchical linear regression models are used to estimate the treatment effects (with 95% CIs), and are adjusted for important patient-level covariates. These will be defined in the final approved statistical analysis plan which will include specific methods of analysis for all outcome variables. We have included estimation of and adjustment for group effects. If there is negligible group effect, then the usual linear regression will be used for the analysis. Any categorical data is assessed in a similar way, using logistic regression models. Prespecified subgroup analyses examine the interaction of treatment assignment with symptoms of anxiety/depression and baseline opioid use. Analysis is conducted using formal tests of interaction.41 This trial is not powered to identify interactions. Thus, while prespecified, these analyses should be considered as no more than exploratory. We are exploring the extent to which change in opioid use, or changes in self-efficacy, mediate change in activities of daily living to gain some understanding as to whether any effects seen are the non-specific effects of the behavioural component of the intervention or they are specifically due to change in opioid usage.
Health economic evaluation
Published evidence and data from the COping with persistent Pain, Effectiveness Research in Self-management (COPERS) study,23 informed the process of conceptualising the structure of a decision analytic model — representing the treatment pathway of individual’s on long-term opioid therapy for non-malignant chronic pain. Data requirements to populate our model structure were used to inform the data collection strategy of the main I-WOTCH trial. The economic analysis of the I-WOTCH study will be in three stages. First, published evidence and individual patient level data from the I-WOTCH internal pilot and COPERS studies will be used to populate the model structure and conduct a Bayesian value of information analysis to identify those parameters for which additional data collection is warranted. Second, we will conduct a within-trial cost-consequences analysis from the perspective of the NHS and social services. Third, a model-based cost-utility analysis will be conducted to estimate the long-term cost-effectiveness of the I-WOTCH intervention versus best usual care. This comprehensive iterative approach has been tested and successfully implemented by one of the applicants in the context of a number of previous National Institute Health Research42 and Medical Research Council (MRC) funded studies.43 44
Process evaluation and intervention fidelity
The process evaluation will investigate any barriers and enablers to the intervention recommendations becoming part of everyday behaviour patterns, from both the perspective of those delivering and receiving the intervention. We will collect observational data by digitally audio-recording all intervention interactions. We will analyse 10% of the recordings to assess fidelity to protocol and further investigate interaction between facilitators and participants. Process evaluation includes outcomes around motivation, expectation and confidence in ability to reduce opioids.
We will conduct semi-structured interviews with approximately 20 participants in the intervention and 20 in the control arms. To ensure a diverse range of views, participants will be selected purposively by age, gender, geographical location, baseline and follow-up opioid use. We will also undertake interviews with a sample of staff delivering the intervention about their experiences of teaching it including enablers and barriers. More information can be found in the protocol for the process evaluation.45
End of trial
The end of the trial is defined as the date when the last participant completes their 12 month follow-up after randomisation. However, follow-up data collection will proceed beyond this date, in particular interviews with participants contributing to the process evaluation.
Patient and public involvement
Two lay advisers with chronic pain, withdrawal of opioids and substantive experience of clinical trial research have been fully involved in the development of the study and intervention. One remains an active member of the study team (CT), the other took retirement from her role during the study (SB). Both lay advisors were recruited via Universities/User Teaching and Research Action Partnership (UNTRAP).
Additionally, prior to receiving funding for the study we ran two meetings at the North East and North Cumbria Clinical Research Network. The meetings included volunteers (n=19) who were people living with chronic non-malignant pain, some of whom had discontinued opioids without medical supervision, others who had discontinued with GP or pain clinic supervision. Both events allowed discussion and input into the design of the intervention structure; (group days and one-to-one support) length of the intervention and content to be covered based on education, motivation, support and providing alternative pain management techniques. The meetings also allowed discussion of the design of the study which included randomisation, best usual care intervention, recruitment processes as well as outcome measures to collect. Patient and public involvement (PPI) participants did not feel the intervention or outcome measures were burdensome and welcomed both arms of the intervention as support for opioid tapering. They also supported the idea of having a lay person with chronic pain with experience of opioid withdrawal to deliver the group days in the active intervention alongside a clinician. In addition to the PPI events, and again prior to receiving funding for the study, we were able to pilot the facilitator training and delivery of the I-WOTCH intervention as part of the Hambleton and Richmond, clinical commissioning group funded community pain service in the North East. This allowed feedback into what worked well and recommendations for changes and improvements.
We will disseminate findings of the main study for the patient participants and group facilitators through a study newsletter and post a lay summary on the study website. In partnership with our PPI representatives we will also feedback to the organisations they represent such as UNTRAP and the PPI events as part of the North East and North Cumbria Clinical Research Network.
Ethics and dissemination
The University of Warwick (Research Impact Services, University of Warwick, Coventry CV4 7AL) is the Sponsor for the study. The study is being conducted in full adherence with the principles of the Declaration of Helsinki and MRC Good Clinical Practice principles and guidelines. It also complies with all applicable UK legislation and Warwick Standard Operating Procedures. All data are being stored securely and held in accordance with the Data Protection Act 2018. All identifiable data are pseudo-anonymised and treated as confidential. Patients have the choice of whether or not to participate and are given all relevant information about the study to make an informed decision. Participants are informed that they are free to withdraw from the trial at any time during any phase of the work without providing a reason and without prejudice, if they so wish. The findings will be disseminated in peer-reviewed journals. We will also publish results on the study website and produce a newsletter for the study facilitators and patient participants. We will engage with NHS organisations, managers, policymakers and clinical commissioning groups to ensure effective dissemination of the findings and inform national, and international, guidance on opioid reduction in this population. Appropriate local approvals were sought for each area in which recruitment was undertaken. The trial is being co-ordinated by the WCTU, University of Warwick.
Acknowledgments
We would like to thank Ms Sally Brown for her valuable input in the early stages of the study. We would also like to thank all of our PPI volunteers and the North East and North Cumbria clinical research network for hosting the events. We would like to thank our clinical and lay facilitators who delivered the intervention and the Clinical Research Networks who assisted in recruitment for the study (North East & North Cumbria, West Midlands, East Midlands, Thames Valley & South Midlands). We would also like to thank Dr Alison Hipwell for her contribution to the intervention development and Dr Celia Bernstein for her contribution to the I-WOTCH study.
References
Footnotes
Contributors All authors read and approved the manuscript. All authors have contriubuted to study design. HS and SE are Co-Chief Investigators and oversee the running of the study. MU has provided input into all aspects of the study design and support in running of the study. CT has provided input into intervention development and delivery as well all study material. CA, KS and VN are leading the process evaluation. AM and CPIU are leading the health economics modeling and formulation. RL, KB and DM have developed the statistical analysis plan and provide statistical support for the study. ST, DC, JN, NT, AF and JS have provided input into the design of the I-WOTCH intervention and training of facilitators. SB, ST and AR have provided input into the design of the study and provide clinical expertise. KH has provided input into study design with particular focus on outcome measures. SA, LB are trial managers and oversee the day to day running of the study. EW provides senior project management to the study.
Funding This project is funded by the National Institute for Health Research (46), Health Technology Assessment (HTA) (project number 14/224/04). The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the HTA, NIHR, NHS or the Department of Health.
Competing interests MU was Chair of the NICE accreditation advisory committee until March 2017 for which he received a fee. He is the chief investigator or co-investigator on multiple previous and current research grants from the UK National Institute for Health Research, Arthritis Research UK and is co-investigator on grants funded by the Australian NHMRC. He is an NIHR Senior Investigator. He has received travel expenses for speaking at conferences from the professional organisations hosting the conferences. He is a director and shareholder of Clinvivo Ltd that provides electronic data collection for health services research. He is part of an academic partnership with Serco Ltd related to return to work initiatives. He is a co-investigator on a study receiving support in kind from Orthospace Ltd. He is an editor of the NIHR journal series, and a member of the NIHR Journal Editors Group, for which he receives a fee. SE is investigator on a number of NIHR and industry sponsored studies. He received travel expenses for speaking at conferences from the professional organisations. SE consults for Medtronic, Abbott, Boston Scientific and Mainstay Medical, none in relation to opioids. SE is chair of the BPS Science and Research Committee. SE is deputy Chair of the NIHR CRN Anaesthesia Pain and Perioperative Medicine National Specialty Group. SE’s department has received fellowship funding from Medtronic as well as nurse funding from Abbott. HS is director of Health Psychology Services Ltd, providing psychological services for a range of health related conditions. AF developed an app that is sold in iTunes for US$9.99 (Opioid Manager). The app is owned by the hospital (UHN) where Dr Furlan works, and Dr Furlan does not retain any profits of the sales of this app for herself. KS received grant funding as PI and CoI from NIHR for other projects. She was on the NIHR HS&DR Funding Board until January 2018. NT received grant funding as PI and CoI from NIHR for other projects and current funding as PI from the Medical Research Council.
Ethics approval Full approval was given by Yorkshire & The Humber - South Yorkshire Research Ethics Committee on 13 September, 2016 (16 /YH/0325).
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.