Article Text

Abstract
Introduction Hepatocyte nuclear factor 1α (HNF1A)-diabetes is the most common monogenetic subtype of diabetes. Strict glycaemic control is crucial for a good prognosis for patients with HNF1A-diabetes. Sulfonylurea (SU) is used as a first-line therapy in HNF1A-diabetes. However, SU therapy may be problematic as it confers a high risk of hypoglycaemia. We hypothesise that low dose of SU in combination with a dipeptidyl peptidase 4 inhibitor provides a safer and more efficacious treatment in patients with HNF1A-diabetes compared with SU as monotherapy.
Methods and analysis In a randomised, double-blinded, crossover study, patients with HNF1A-diabetes will randomly be assigned to 16 weeks of treatment with glimepiride+linagliptin, 4 weeks of washout and 16 weeks of treatment with glimepiride+placebo (or vice versa). Treatment will be evaluated with continuous glucose monitoring and combined meal and bicycle tests conducted at baseline and at the end of each of the two treatment periods. The primary end point is the absolute difference in the mean amplitude of glycaemic excursions between the two treatments (glimepiride+linagliptin vs glimepiride+placebo) at the end of each treatment period.
Ethics and dissemination The study protocol is approved by the Danish Medicines Agency, The Scientific-Ethical Committee of the Capital Region of Denmark (H-17014518) and the Danish Data Protection Agency. The trial will be carried out and monitored in compliance with Good Clinical Practice guidelines and in accordance with the latest version of the Declaration of Helsinki. Positive, negative and inconclusive results will be published at scientific conferences and as one or more scientific manuscripts in peer-reviewed journals with authorship in accordance with the International Committee of Medical Journal Editors’ recommendations.
Trial registration number 2017-000204-15.
- general diabetes
- genetics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study is the first randomised, double-blinded, placebo-controlled, crossover trial, which evaluate combination therapy of glimepiride+linagliptin in patients with hepatocyte nuclear factor 1α (HNF1A)-diabetes.
Treatment will be assessed by continuous glucose monitoring, which provides a comprehensive overview of glucose fluctuations in addition to haemoglobin A1c and patient-reported outcomes including episodes of hypoglycaemia.
Treatment effects on endocrine pancreas function and hypoglycaemic response will be thoroughly evaluated during a combined meal and bicycle test.
Due to the relatively small sample size, secondary outcome measures might not be generalisable to all patients with HNF1A-diabetes.
Introduction
Diabetes is a global health challenge affecting >400 million patients worldwide.1 A minimum of 1% of all cases of diabetes is due to heritable monogenetic subtypes of diabetes collectively named maturity-onset diabetes of the young (MODY).2 The majority of patients diagnosed with MODY have a mutation in the hepatocyte nuclear factor 1α (HNF1A) gene, a subtype of MODY termed MODY type 3 or HNF1A-diabetes.3 4 The MODY diagnosis should be considered when an individual displays: (1) hyperglycaemia, (2) autosomal dominant inheritance of diabetes (diabetes in at least two consecutive generations), (3) early onset of diabetes (at least one family member with onset before 25 years of age), (4) measurable C-peptide levels in plasma 3 years after diagnosis and (5) lack of autoantibodies against pancreatic islet cells.5 The risk of microvascular complications (neuropathy, nephropathy and retinopathy) and macrovascular complications (eg, stroke, myocardial infarction and peripheral artery disease) in HNF1A-diabetes is similarly high to that of patients with type 2 diabetes.6 Thus, strict glycaemic control combined with reduction of other cardiovascular risk factors and proper treatment of complications are crucial for a good prognosis.6 7
A key characteristic of HNF1A-diabetes is a reduced glucose-induced insulin response.8 In the insulin-secreting beta cells, the turnover of glucose (glycolysis) is impaired. This results in reduced intracellular levels of adenosine triphosphate (ATP). Decreased ATP levels impede ATP-dependent potassium channel (KATP-channel) essential to exocytosis of insulin. Sulfonylurea (SU) binds to SU receptor 1 (SUR1), a subunit of the KATP-channel, hereby closing the channel. This causes membrane depolarisation leading to opening of the voltage-gated calcium (Ca2+) channel and increasing intracellular Ca2+ concentration, which in turn causes docking of secretory vacuoles and exocytosis of insulin (ie, insulin secretion).9 10 Due to a high sensitivity to SUs combined with normal or even increased insulin sensitivity, this treatment is highly effective with respect to lowering of plasma glucose.
Thus, SU treatment is currently the recommended first-line therapy in patients with HNF1A-diabetes and is in general well-tolerated and considered safe.11 12 However, due to the glucose-independent action of SUs, they confer a risk of hypoglycaemia even when relatively low doses are used.12 13 In 2006, Tuomi et al 14 demonstrated that 40% of their patients with HNF1A-diabetes developed hypoglycaemia during physical exercise (ie, light cycling for 30 min 2 hours after meal ingestion) when treated with SU (glibenclamide). One patient experienced prolonged hypoglycaemia for 12 hours. In a clinical setting, some patients with HNF1A-diabetes have recurrent episodes of hypoglycaemia on SU treatment. In these patients, a reduction of SU dosing may protect against hypoglycaemia; however, this may be at the expense of good glycaemic control. Therefore, patients with HNF1A-diabetes with recurrent hypoglycaemia on SU treatment and unsatisfactory haemoglobin A1c (HbA1c) levels may benefit of a more effective treatment with a lower risk of hypoglycaemia.
In healthy individuals, the incretin hormones glucose-dependent insulinotropic peptide (GIP) and glucagon-like peptide-1 (GLP-1) are important regulators of plasma glucose due to their effects on insulin and glucagon secretion. GIP and GLP-1 are secreted from enteroendocrine cells in the intestinal epithelium.15 The major secretory stimulus is the presence of nutrients in the gut lumen.16 GLP-1 exerts potent insulinotropic and glucagonostatic effects in a glucose-dependent fashion; that is, the insulin-stimulating and glucagon-suppressive effects only are active during elevated plasma glucose levels (>4–5 mmol/L). GIP displays a similar glucose-dependent insulinotropic effect as GLP-1 in healthy adults, but it does not exert any glucagon-suppressive effect; in fact, GIP has been shown to stimulate glucagon secretion during euglycaemia or hypoglycaemia.17 This glucose-dependency makes the incretin hormones essential as ‘glucose-stabilisers’ that decrease high glucose levels and elevate glucose levels during hypoglycaemia.18 In HNF1A-diabetes incretin hormones are secreted in a normal fashion, but the insulinotropic effect of GIP and GLP-1, respectively, is diminished.19 Both incretin hormones are degraded rapidly by the ubiquitous enzyme dipeptidyl peptidase 4 (DPP-4). Patients with HNF1A-diabetes display increased DPP-4 activity during fasting, but the significance of this finding is unknown.19 Incretin-based drugs are increasingly being used for the treatment of patients with type 2 diabetes: (1) DPP-4 inhibitors prevent breakdown of endogenously secreted incretin hormones and (2) injectable DPP-4 resistant GLP-1 receptor agonists (GLP-1RA) result in supraphysiological levels of circulating GLP-1RA.20 21 In a recent randomised controlled trial, we compared treatment with SU and GLP-1RA in HNF1A-diabetes and demonstrated significant reductions in fasting plasma glucose and postprandial glucose excursions in patients with HNF1A-diabetes with both treatments.12 The glucose-lowering effect was greater with SU, but at the expense of a markedly higher risk of hypoglycaemia.
Hypothesis and aim
We hypothesise that a low dose of glimepiride (a SU compound) in combination with linagliptin (a DPP-4 inhibitor) will provide an efficacious and safe treatment with a better glycaemic control and lower risk of hypoglycaemia compared with glimepiride as monotherapy. The rationale for combination therapy is: (1) DPP-4 inhibitors will display a glucose-dependent insulinotropic effect elicited by increased concentrations of active GIP and GLP-1; (2) DPP-4 inhibitors may protect against hypoglycaemia due to the added effects by GIP on glucagon secretion; (3) the small dose of SU will compensate for the decreased ATP levels and the malfunction of KATP in the insulin-secreting beta cells; (4) SU given in a low dose (in combination with a DPP-4 inhibitor) will protect against hypoglycaemia.
Study objective
The objective of this study is to investigate the efficacy and safety of combination therapy with glimepiride and linagliptin compared with glimepiride monotherapy in patients with HNF1A-diabetes. The primary end point is the difference in glycaemic variability defined as mean amplitude of glycaemic excursions (MAGE) between the two treatment arms at end of treatment. MAGE has been chosen as most patients with HNF1A-diabetes have only mildly deranged fasting plasma glucose values and marked postprandial hyperglycaemia. An improvement in MAGE corresponds with an improvement of postprandial hyperglycaemia. Secondary end points consist of several parameters evaluating glycaemic variability, long-term glycaemic control and hypoglycaemia.
Methods and analysis
Study design
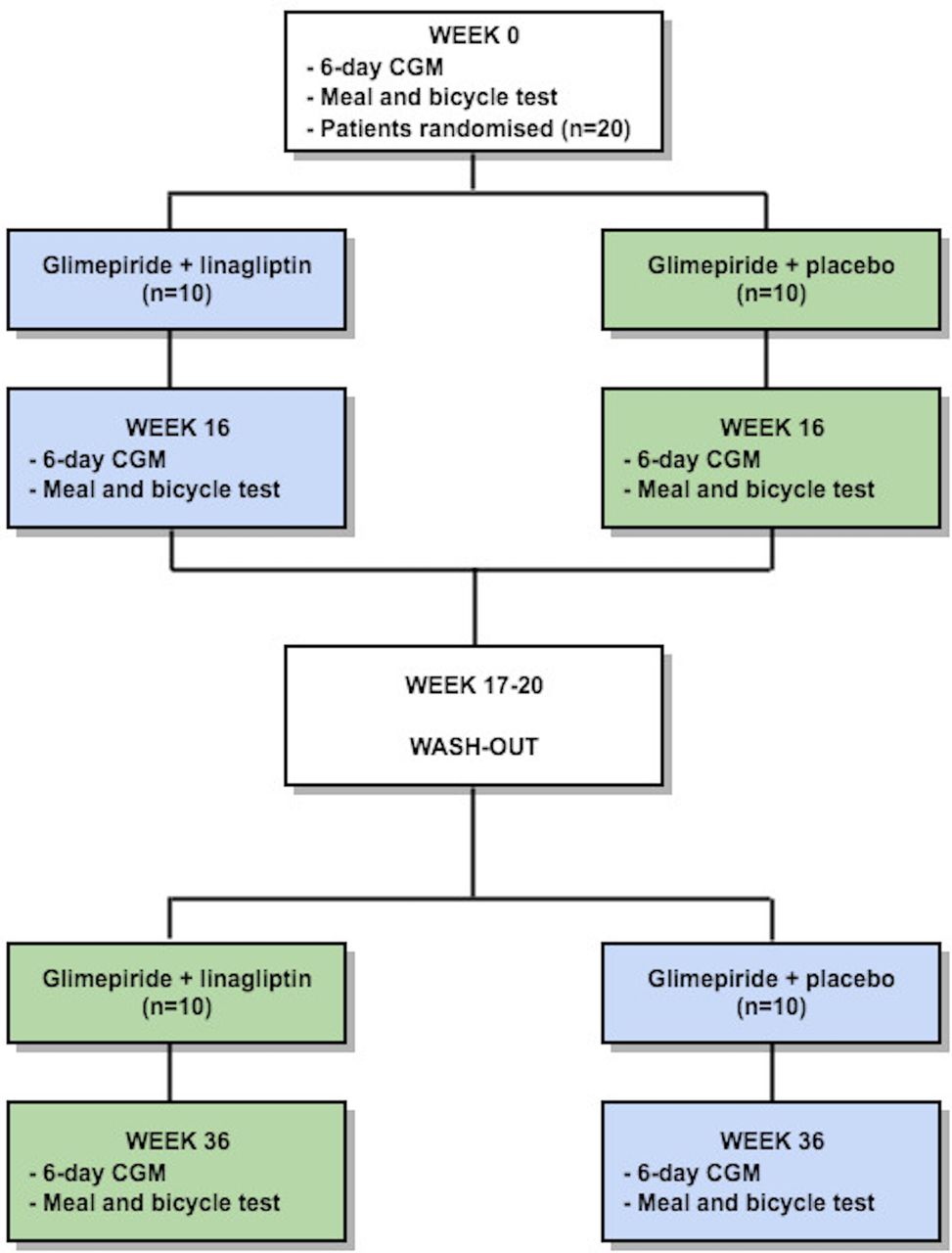
This trial is a single-centre, randomised, double-blinded, crossover study in patients with HNF1A-diabetes (figure 1). The study is intended to show superiority of glimepiride+linagliptin in regard to the primary end point.

Trial design. Patients with hepatocyte nuclear factor 1α-diabetes will be randomly assigned to one of two double-blinded treatment sequences: (1) 16 weeks of treatment with glimepiride+linagliptin, 4 weeks of washout and 16 weeks of treatment with glimepiride+placebo (blue boxes); and (2) 16 weeks of treatment with glimepiride+placebo, 4 weeks of washout and 16 weeks of treatment with glimepiride+linagliptin (green boxes). Evaluation of the glycaemic variability will be assessed by continuous glucose monitoring (CGM) and meal and bicycle test in weeks 0, 16 and 36. For other investigations during the study, see text for more details.
Study participants
A total of 20 patients with HNF1A-diabetes will be enrolled. Patients will be recruited from the diabetes outpatient clinics at Gentofte Hospital and Steno Diabetes Center Copenhagen, Denmark or by letter to patients registered in a database at Steno Diabetes Center Copenhagen. All trial activities will be executed at Clinical Metabolic Physiology, Steno Diabetes Center Copenhagen, Gentofte Hospital, Hellerup, Denmark.
The total number of participants in the following categories will be recorded: invited, excluded before screening (including reasons for exclusion), declined to participate, not reached, screened, excluded at screening, randomised, completed intervention period, withdrew/dropped out during the study period (including reason for withdrawal/dropping out), lost to follow-up and analysed. Eligible participants will receive written and oral information about study. Sufficient time for reflection will be given before written informed consent is obtained as according to The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Good Clinical Practice (GCP) guidelines.
Eligibility of participants
Inclusion criteria
Patients with HNF1A-diabetes caused by heterozygous mutation in HNF1A confirmed by Sanger sequencing of the gene.
Monotherapy with diet or a stable dose of glimepiride of (≥0.5 mg/day) during 4 weeks.
Patients on glimepiride treatment: HbA1c≥6.5% (HbA1c≥48 mmol/mol).
Patients receiving no glucose-lowering agents: HbA1c≥7.0% (HbA1c≥53 mmol/mol).
Age≥18 years of age.
Capability to perform a 30 min light bicycle test at a heart rate of 100–120 beats per min.
Fertile females must use adequate contraceptive method (intrauterine contraceptive devices or hormonal contraception).
Informed consent.
Exclusion criteria
Use of glucose-lowering drugs other than glimepiride.
Uraemia, end-stage renal disease or estimated glomerular filtration rate<30 mL/min/1.73 m2 and/or albuminuria.
Liver disease and/or serum alanine aminotransferase (ALT) and/or serum aspartate aminotransferase (AST)>2× upper normal serum levels.
Anaemia (males blood haemoglobin <8.0 mmol/L and females <7.0 mmol/L).
History of acute and/or chronic pancreatitis.
Pregnancy or breast feeding.
Inability to complete the study.
Known allergic reaction to study medication.
Intention to become pregnant.
Intervention
Participants will be randomly assigned to one of two treatment sequences: (1) 16 weeks of treatment with glimepiride+linagliptin, 4 weeks of washout and 16 weeks of treatment with glimepiride+placebo or (2) 16 weeks of treatment with glimepiride+placebo, 4 weeks of washout and 16 weeks of treatment with glimepiride+linagliptin. Patients treated with glimepiride at screening will continue on the same dosage after randomisation. Treatment-naïve patients will start on 0.5 mg glimepiride after randomisation. Each treatment period is divided into a drug-titration period (weeks 1–4) and a maintenance period (weeks 5–16). After initiation of linagliptin/placebo patients will continue on the initial dose of glimepiride at screening for 1 week. Patients are instructed to measure fasting plasma glucose values daily. After the first week of linagliptin/placebo, the glimepiride dose will be adjusted for the first time. Target fasting plasma glucose (average during at least 5 days) is between 4.5 and 6.0 mmol/L (both inclusive) without episodes of symptomatic or biochemical hypoglycaemia (plasma glucose≤3.9 mmol/L). In case of no hypoglycaemia and an average fasting plasma glucose>6 mmol/L, the glimepiride dose will be increased with 0.5 mg. In case of hypoglycaemia or average fasting plasma glucose <4.5 mmol/L, the dose of glimepiride will be decreased by 0.5 mg. In the remaining titration period, the glimepiride dose will be uptitrated or downtitrated once-weekly in the same treat-to-target manner with a maximum daily dose of 6 mg in both groups. A total daily dose of glimepiride≥1 mg will be divided and administered as two daily doses: one dose in the morning and one dose in the evening. If target fasting plasma glucose is not achieved during the drug-titration period, the dose of glimepiride will be adjusted after week 4 at the investigators discretion. In case of hypoglycaemia outside of the drug-titration period, glimepiride dose will be down-titrated 0.5 mg. Once-daily linagliptin 5 mg or placebo will be initiated at each treatment period and the dose kept stable throughout the experiment. After the first treatment period, the dose of glimepiride at randomisation will immediately be reintroduced for all patients and followed by a 4-week ‘linagliptin/placebo’ washout period. Participants will be encouraged not to change their lifestyle (ie, diet, exercise and smoking status) during the trial. There will be no restriction to concomitant non-antidiabetic medications. Trial medicine will comply with Good Manufacturing Practice. Drug accountability will be performed at all clinical visits to document degree of compliance.
Outcomes
Primary outcome measurements
The primary end point is the absolute difference in MAGE between the two treatments (glimepiride+linagliptin vs glimepiride+placebo) at the end of each treatment period calculated from 6 days (144 hours) of continuous glucose monitoring (CGM).
Secondary outcome measurements
Secondary end points include differences between the two treatments (glimepiride+linagliptin vs glimepiride+placebo) in:
Other parameters of glycaemic variability including low blood glucose index (LBGI), SD of mean glucose, time spent in hypoglycaemia and time spent in hyperglycaemia calculated from 6 days of CGM at the end of each treatment period.
Difference between baseline and end of treatment in parameters of glycaemic variability including MAGE, LBGI, SD of mean glucose, time spent in hypoglycaemia, time spent in hyperglycaemia from 6 days of CGM.
The absolute glycaemic excursion during meal and bicycle test.
Change in pancreatic endocrine function during meal and bicycle tests (insulin, C-peptide and glucagon).
Change in counter-regulatory responses to hypoglycaemia during meal and bicycle tests (growth hormone (GH), cortisol and norepinephrine).
Number and severity of hypoglycaemic events during meal and bicycle tests.
Number and severity of hypoglycaemic events during treatment periods.
Fasting plasma glucose at the end of each treatment period.
HbA1c at end of trial.
Fructosamine at end of trial.
Changes in bodyweight from baseline.
Changes in cardiovascular biomarkers in urine.
Changes in quality of life from baseline evaluated with Short Form-36.
Dose of glimepiride at the end of each treatment period.
Differences between treatments in the proportion of patients achieving HbA1c<6.5% (HbA1c<48 mmol/mol) (or a decrease in HbA1c>5 mmol/mol compared with baseline) and have no episodes of hypoglycaemia during the study.
The outcome parameters are defined in box 1.
Definition of outcomes
MAGE is a measure of glycaemic variability. It quantifies the mean amplitude of all glycaemic excursions that exceed 1 SD of the plasma glucose within the last 24 hours. High MAGE values are strongly associated with hyperglycaemia.
LBGI is a measure of glycaemic variability. It describes the frequency and extent of hypoglycaemia. High values of LBGI are correlated with high risk of severe hypoglycaemia.
Other measures of glycaemic variability will include SD of mean glucose, time spent in hypoglycaemia and time spent in hyperglycaemia.
The absolute glycaemic excursion evaluated as the absolute difference from the highest and lowest plasma glucose measurement during meal and bicycle test.
Pancreatic endocrine function evaluated as AUC for plasma insulin, C-peptide and glucagon during meal and bicycle test.
Counter-regulatory responses to hypoglycaemia evaluated as AUC for plasma GH, cortisol and norepinephrine during meal and bicycle test.
Number and severity of hypoglycaemic events for each treatment period will be reported by patients in diaries. Hypoglycaemic events will be described according to the American Diabetes Association’s recommendations.
Number and severity of hypoglycaemic events during meal and bicycle test will be measured as defined in the bullet point above.
Fasting plasma glucose defined as plasma glucose measurement taken after at least an 10 hours fast.
HbA1c is an indicator for time-averaged long-term plasma glucose levels and thus glycaemic control over approximately 90–120 days.
Fructosamine is an indicator for time-averaged plasma glucose levels over 2–3 weeks. It reflects the total amount of glycated plasma proteins.
Bodyweight will be measured before and after treatment in both arms at the day of meal and bicycle test.
Changes in the cardiovascular biomarker 8-oxoGuo: 8-oxoGuo is a urinary marker of oxidative stress. 8-oxoGuo is produced when free radicals oxidise ribonucleic acids. Increased urinary excretion of 8-oxoGuo has been associated with increased mortality in newly diagnosed type 2 diabetes patients and after 6 years.
Changes in quality of lifeevaluated with SF-36: SF-36 is a validated questionnaire for health-related quality of life measurement in type 2 diabetes. A decrease in the score in SF-36 have been linked to increased risk of hospitalisation and mortality.
Dose of glimepiride at the end of each treatment period as total daily dose of glimepiride.
Differences between treatments in the proportion of patients achieving HbA1c<48 mmol/mol (or a decrease in HbA1c>5 mmol/mol compared with baseline) and have no episodes of hypoglycaemia during the study.
8-oxoGuo, 8-oxo-7,8-dihydroguanosiene; AUC, area under the curve; GH, growth hormone; HbA1c, haemoglobin A1c; LBGI, low blood glucose index; MAGE, mean amplitude of glycaemic excursions; SF-36, Short Form 36.
Participant timeline
Participant timeline is illustrated in table 1. Study treatment will be evaluated with clinical visits and blood samplings, telephone calls, CGM and meal and bicycle test. The full study period constitutes of 36 weeks (one washout period of 4 weeks and two treatment periods of 16 weeks), preceded by a pretreatment evaluation (screening) approximately 14 days before randomisation.
Participant timeline
Screening
Participants will be screened according to inclusion and exclusion criteria. Demographics and medical history will be obtained, including recording of current medication and previous tests verifying the diagnosis of HNF1A-diabetes. Vital signs (blood pressure and pulse), height and weight, waist and hip circumference will be measured and a physical examination will be performed. Screening blood samples will be taken including haemoglobin, creatinine, electrolytes (Na+, K+), C reactive protein (CRP), thyroid-stimulating hormone (TSH), ALT, AST, bilirubin, alkaline phosphatase, albumin, plasma glucose, HbA1c, urine examined for albumin:creatinine ratio and cardiovascular biomarkers. Fertile women will undergo a pregnancy test (urine sample). If the potential participant meets all inclusion criteria and none of the exclusion criteria, an appointment for the following visits (randomisation) will be set up. For screening failures (patients who were screened, but not randomised), a screening failure form will be completed.
Continuous glucose monitoring
Blinded CGM measurements will be conducted over 6 days three times during the trial at baseline and after each treatment period (weeks 0, 16 and 36). Urine samples will be collected daily during CGM.
Telephone contacts
During the 36 weeks, a total of six telephone calls will be conducted. At these contacts, the glimepiride treatment will be titrated as described previously according to patients’ diaries, reported hypoglycaemic episodes and adverse events (AE). Compliance will be noted. Patients will be informed to continue on 5 mg of linagliptin/placebo forward. The telephone contacts will be done in the drug-titration periods (weeks 1, 2, 3, 21, 22 and 23).
Clinical visits and blood sampling
Patients will meet for eight clinical visits. Compliance and AEs will be noted. At clinical visits dispensing of trial drugs will be done. Blood samples will be performed during clinical visits and at the end of study. The following analysis will be performed: haemoglobin, vitamin b12, creatinine, electrolytes (Na+, K+), leucocytes, thrombocytes, CRP, C-peptide, cholesterols, triglycerides, TSH, CRP, bilirubin, ALT, AST, alkaline phosphatase, albumin, coagulation factors, plasma glucose, HbA1c and fructosamine. Visits are scheduled for weeks 4, 8, 12, 16, 24, 28, 32 and 36.
Meal and bicycle test
Three meal and bicycle tests (figure 2) will be performed at baseline and in the end of each of the two treatment periods (weeks 0, 16 and 36). Patients will be subjected to a standard 4-hour liquid meal test and a concurrent 30 min bicycle test. The patients will meet in a fasting (10 hours) state. Patients will continue their therapy according to trial protocol and thus take glimepiride tablets as normal the evening before, and glimepiride and linagliptin/placebo tablets at the clinic 1 hour prior to the ingestion of the meal (time −60 min). Patients will ingest a standard liquid meal (525 kcal: 65 g carbohydrate, 20 g fat, 21 g protein) added 1.5 g of paracetamol at time 0 min. From 60 to 90 min, a 30 min bicycle test will be performed (initially 50–60 rotations per minute, with adjustment of bicycle resistance every 5 min until achieving a heart rate of 90–110 beats per min). Symptoms of hypoglycaemia will be monitored and plasma glucose measured on prespecified time points and in the event of symptoms of hypoglycaemia. Other AEs and compliance will also be noted. During the meal test, blood samples will be drawn at prespecified time points for measurement of plasma glucose, insulin, C-peptide, glucagon, GIP, GLP-1, paracetamol and counter-regulatory hormones (GH, cortisol, norepinephrine).

{kind=link}
{kind=link}
Meal and bicycle test. Participants arrive fasting. Study medication is taken at time −60 min. At time 0 min a mixed liquid meal (525 kcal) added 1.5 g of paracetamol is served. From 60 to 90 min, a light bicycle test is performed (target heart rate 90–110 beats per min). Blood samples are taken for the analysis of plasma glucose, insulin, C-peptide, glucagon, incretins and counter-regulatory hormones. Patients will be monitored for any symptoms of hypoglycaemia. FPG, fasting plasma glucose.
Sample size
The study is a superiority study which will estimate the effect of two different treatments with the hypothesis that the effect of treatment with glimepiride+linagliptin versus glimepiride+placebo will display a significant decrease in MAGE.
For power calculation on our primary end point, the following equation has been used as recommended by Jones and Kenward22:
N = (Zα+Zβ)2×SD2/MIREDIF2
Zα is a table value according to a two-sided standard normal distribution;
Zβ is a table value according to a one-sided standard normal distribution;
SD is the standard deviation;
MIREDIF is the minimum relevant difference.
The power of our study (1−β) will be set at 0.8, and the risk of accepting a false hypothesis is 0.20 (β=0.20) with a significance level at 0.05 (α=0.05). Thus, Zα=1.96 and Zβ=0.84. We have defined a minimal relevant difference (MIREDIF) of 2 mmol/L (36 mg/dL) in MAGE between the two groups. Power calculation in the current trial has been based on the SD (SD=50) in MAGE presented by Saisho et al.23
Number of participants needed:
N=(1.96+0.84)2×502/362=15.1≈16
In order to allow drop-outs, a total of 20 patients with HNF1A-diabetes will be randomised. With the chosen size sample a risk of type 2 error exists, thus missing an effect of glimepiride+linagliptin versus glimepiride placebo. However, we consider only an effect that can be found within a sample size of 20 patients to be clinically relevant.
Assignment of interventions
Patients will be randomised in 1:1 allocation using a computer-generated random list of numbers. The list of random numbers will be made by an employee who is otherwise uninvolved in the study. The allocation will not be known to the investigators or patients during the study. Everyone involved in the trial will be blinded until data are fully analysed. Code break of a participant’s trial medicine will occur during the trial only in case of exceptional circumstances when knowledge of treatment is absolutely necessary. Only the sponsor-investigator and an employee who is otherwise uninvolved in the study will be able to perform a code break.
Data collection, analysis and management
All data will be collected in electronic case report forms in Research Electronic Data Capture (REDCap) hosted at the Capital Region of Denmark. REDCap is a secure, web-based application designed to support data capture for research studies, providing: (1) an intuitive interface for validated data entry; (2) audit trails for tracking data manipulation and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages and (4) procedures for importing data from external sources.24 The case report forms will only be available for investigators and monitors according to ICH-GCP guidelines.25 The database will be locked database after last participant’s end-of-study visit. Only investigators will have access to the final data set. To ensure adherence to protocol, a designated trial project coordinator will be responsible for reminding patients of dates for visits and clinical tests. Additionally, drug accountability will be performed to ensure compliance. CGM monitors used will be the iPro2 CGM from Medtronic, Northridge, California, USA, which has been validated elsewhere.26 To ensure data collection, only CGM-trained investigators will assemble and disassemble the CGM monitor at the study site. Investigators will receive training by Medtronic representatives before commencement of the trial. All data are stored electronically by the CGM device and uploaded to a server hosted by Medtronic. If participants have any problems with their CGM monitor, an investigator will always be accessible by phone and provide guidance. If a problem with the CGM monitor persists, additional clinical visits will be arranged to ensure data collection.
Blood and urine samples from screening visits, clinical visits and meal and bike tests will be analysed in the Department of Biochemistry at Gentofte Hospital or collaborators according to predefined local standards. Additional blood and urine will be stored in the research biobank in case of the need of re-analysis.
Data monitoring
The trial will be monitored by the GCP unit, Bispebjerg Hospital, University of Copenhagen, Copenhagen, Denmark. The GCP unit and the Danish Medicines Agency will be able to conduct an audit of the trial independent of sponsor-investigator and investigators. There will be no data monitoring committee due to (1) the relatively short duration of the study, and because (2) the drugs used in this trial are approved and considered safe. At all telephone contacts and clinical visits, patients’ adverse events, if any, will be noted in the case report forms. Additionally, patients will be encouraged to contact the investigators in case of any AE. Participants will be thoroughly informed of potential side effects when participating in this trial. All AEs will be reported to the Danish Medicines Agency and the Scientific-Ethical Committee of the Capital Region of Denmark at the end of trial. Sponsor-investigator shall be further responsible for reporting of any serious adverse event (SAE), serious adverse reaction (SAR) and suspected unexpected serious adverse reaction (SUSAR), which occur during the conduct of the study to the Scientific-Ethical Committee of the Capital Region of Denmark once yearly in accordance with the applicable laws and regulations. Sponsor-investigator will delegate the responsibility for the expedited regulatory reporting of SAE, SAR and SUSAR to Boehringer Ingelheim. The sponsor-investigator will report every SAE, SAR and SUSAR immediately to Boehringer Ingelheim within 24 hours. Boehringer Ingelheim will be responsible to report SAE, SAR and SUSAR to the Danish Medical Agency in accordance with the applicable laws and regulations.
Statistical analysis
All analyses will be performed on an intent-to-treat sample of subjects who were randomised and received at least one dose of medication or placebo. Continuous data will be summarised by descriptive statistics with the number of observations (n), mean, SD, minimum, median and maximum. Categorical data will be summarised in frequency tables using count and percentages. AUC will be calculated using the trapezoidal rule. Comparisons between treatment outcomes will be performed in linear mixed models for continuous data. Comparisons of binary or ordinal data will be performed in generalised linear models for binary or ordinal data. Missing data will implicitly be imputed by performing likelihood inference in the linear mixed models for continuous data. Interaction between treatment and treatment period will be performed in linear mixed models to test for potential carryover effect. Outcomes that are not normally distributed will be transformed for the analysis in linear mixed models. Not normally distributed outcomes will be presented as median and ranges. All tests will be carried out at a significance level of 5%. AEs will be summarised qualitatively.
Strengths and limitations
The strength of this study is the randomised, placebo-controlled, double-blinded trial design. The crossover design means the patients will be their own control. Glucose-lowering treatment will be evaluated regarding all glucose parameters: glucose variability, long-term glycaemic control (HbA1c) and by the amount of hypoglycaemic events. The duration of each treatment period (16 weeks) will provide time to asses both the efficacy of treatments and risk of hypoglycaemia. The crossover design has the advantage that it reduces the risk of confounding covariates by unbalanced randomisation. However, the disadvantage of a crossover study is if participants change during the study (eg, gain weight) or if there is a carryover effect from one treatment period to another. Given the 4-week washout period the risk of a carryover effect is minimal. The major limitation of this study is the small sample size and rather short duration of the study. Thus, the results of this study might not be generalisable to all patients with HNF1A-diabetes. Another limitation is that participants at randomisation will receive different baseline doses of glimepiride. This may cause different levels of saturation of the SUR1 and thereby potentially interindividual differences to an increment of the glimepiride dose. Third, a limitation of this study is that it might be hard to recruit patients given the criteria for participation and the relatively few diagnosed with HNF1A-diabetes in Denmark.
Patient and public involvement
This study is designed to address the well-known dysregulation and risk of hypoglycaemia in patients with HNF1A-diabetes treated with SU as monotherapy. We expect the study to provide knowledge about the combination therapy with DPP-4 inhibitors and SU, which from a mode of action point of view make a lot of sense. Currently, neither patients nor patient advisers have been involved in the design, recruitment and conduct of the study. All participants will receive a personal treatment evaluation and an evaluation of the whole population in writing after a full data analysis has been performed. The intervention burden will be assessed by the recording of all episodes of AE and/or SAE according to GCP.
Ethics and dissemination
The sponsor-investigator is responsible for informing participants, investigators and the Danish Medicines Agency and The Scientific-Ethical Committee of the Capital Region of Denmark of any amendments to the protocol as per national requirements. The study will be conducted with informed consent from participants according to the Declaration of Helsinki. All data forms and blood samples will be anonymised and only be labelled with the patient’s initial and study number. Sponsor-investigator is responsible for keeping a list separately for all randomised participants containing patient numbers, full names and date of birth. Patients in the study will be covered by insurance via the hospital. Positive, negative and inconclusive results will be published at scientific conferences and as one or more scientific manuscripts in peer-reviewed journals with authorship in accordance with International Committee of Medical Journal Editors.
References
Footnotes
Contributors This study is based on an idea by TV. ASC, HS, SH, TH and FKK contributed to the trial design. ASC wrote the first protocol draft. All authors contributed to the development of the study protocol and approved the final draft. TV is sponsor-investigator. ASC will be responsible for enrolment of patients and operating the trial on a daily basis.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests ASC, SH, TH and TV have nothing to disclose in relation to the present paper. HS reports personal fees from AstraZeneca, personal fees from Boehringer Ingelheim Pharmaceuticals, personal fees from Bristol-Myers Sqiubb, outside the submitted work. FKK reports personal fees from Amgen, grants and personal fees from AstraZeneca, personal fees from Boehringer Ingelheim, personal fees and non-financial support from Eli Lilly, personal fees and non-financial support from MSD/Merck, personal fees and non-financial support from Novo Nordisk, grants, personal fees and non-financial support from Sanofi, grants and personal fees from Zealand Pharma, other from Antag Therapeutics, outside the submitted work.
Patient consent Not required.
Ethics approval The study has been approval by the Danish Medicines Agency, The Scientific-Ethical Committee of the Capital Region of Denmark and the Danish Data Protection Agency.
Provenance and peer review Not commissioned; externally peer reviewed.