Article Text

Abstract
Introduction Acute kidney injury (AKI) occurs in 55–60% of critically ill patients, and sepsis is the most common underlying cause. No pharmacological treatment options are licensed to treat sepsis-associated AKI (SA-AKI); only supportive renal replacement therapy (RRT) is available. One of the limited number of candidate compounds in clinical development to treat SA-AKI is alkaline phosphatase (AP). The renal protective effect of purified bovine intestinal AP has been demonstrated in critically ill sepsis patients. To build on these observations, a human recombinant AP (recAP) was developed, of which safety and efficacy in patients with SA-AKI will be investigated in this trial.
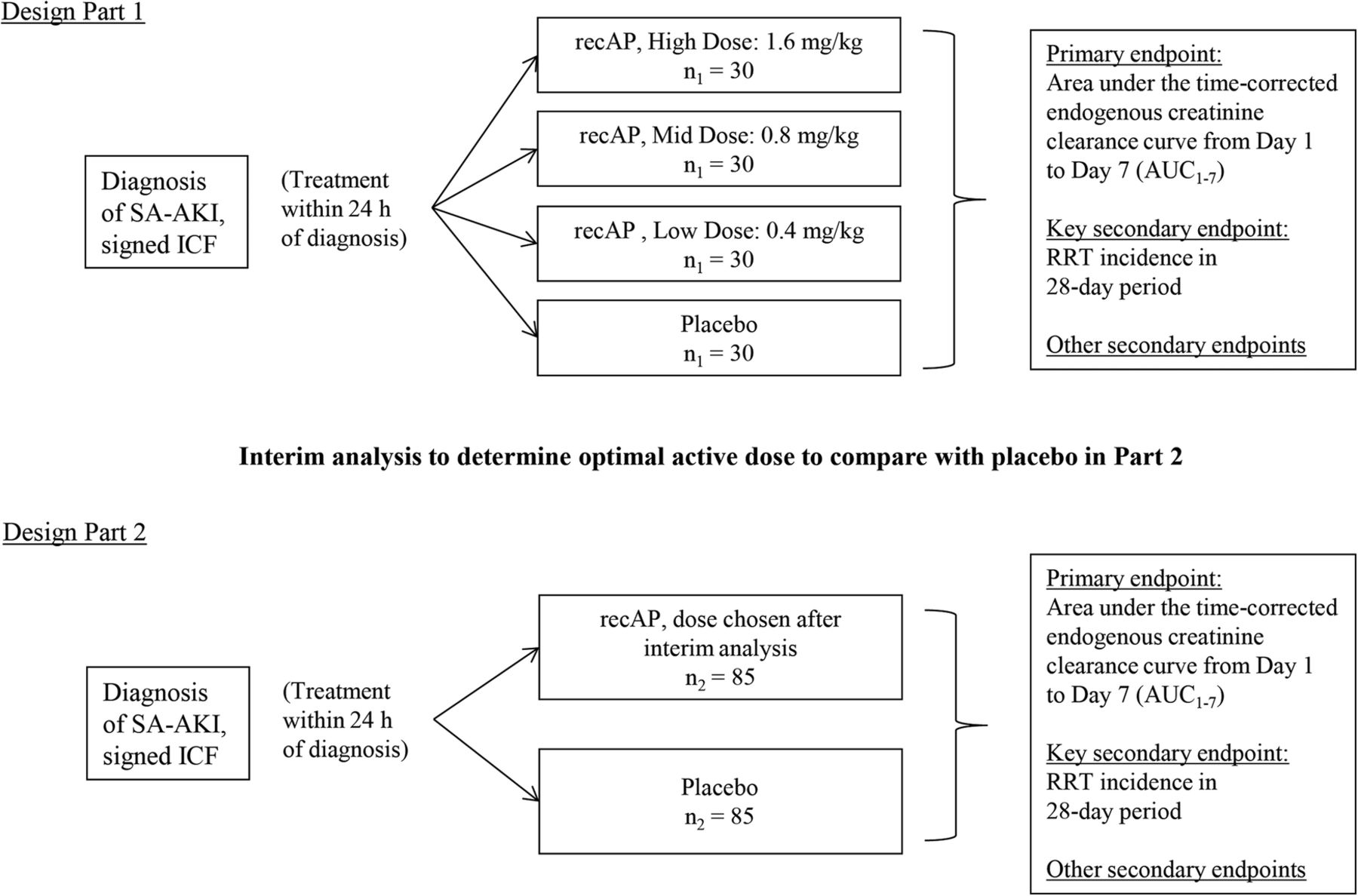
Methods This is a randomised, double-blind, placebo-controlled, 4-arm, proof-of-concept, dose-finding adaptive phase IIa/IIb study, conducted in critically ill patients with SA-AKI. A minimum of 290 patients will be enrolled at ∼50 sites in the European Union and North America. The study involves 2 parts. Patients enrolled during Part 1 will be randomly assigned to receive either placebo (n=30) or 1 of 3 different doses of recAP (n=30 per group) once daily for 3 days (0.4, 0.8 or 1.6 mg/kg). In Part 2, patients will be randomly assigned to receive the most efficacious dose of recAP (n=85), selected during an interim analysis, or placebo (n=85). Treatment must be administered within 24 hours after SA-AKI is first diagnosed and within 96 hours from first diagnosis of sepsis. The primary end point is the area under the time-corrected endogenous creatinine clearance curve from days 1 to 7. The key secondary end point is RRT incidence during days 1–28.
Ethics and dissemination This study is approved by the relevant institutional review boards/independent ethics committees and is conducted in accordance with the ethical principles of the Declaration of Helsinki, guidelines of Good Clinical Practice, Code of Federal Regulations and all other applicable regulations. Results of this study will reveal the efficacy of recAP for the improvement of renal function in critically ill patients with SA-AKI and will be published in a peer-reviewed scientific journal.
Trial registration number NCT02182440; Pre-results.
- sepsis
- acute kidney injury
- randomized controlled trial
- alkaline phosphatase
- therapy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first randomised controlled trial in sepsis-associated acute kidney injury (SA-AKI) with well-controlled comparable standard of care, as participating institutions are required to adhere to the Surviving Sepsis Campaign and KDIGO Clinical Practice Guideline for AKI recommendations and to strictly defined renal replacement therapy starting and stopping criteria.
The study was designed by a group of leading global experts in AKI and sepsis, with input from the US Food and Drug Administration, the European Medicines Agency and several local European country authorities, resulting in a dose-finding adaptive trial.
Results of this trial will allow us to draw conclusions on the efficacy of human recombinant alkaline phosphatase in the improvement of renal function and related clinical parameters.
Regardless of the outcome of the intervention, data from the placebo group will provide valuable information on clinical outcome in patients with SA-AKI.
Strict inclusion criteria and time-windows may limit generalisation of the results to the entire population of critically ill patients with SA-AKI.
Introduction
Acute kidney injury (AKI) is common in critically ill patients and associated with significant morbidity and mortality. The intensive care unit (ICU) incidence of AKI is 55–60% with an associated mortality of more than 27%.1 Patients surviving an AKI episode have an increased risk to develop chronic kidney failure, resulting in a tremendous burden for the patient and society.2 AKI may occur after cardiovascular surgery, administration of nephrotoxic drugs, trauma, burns and sepsis, with the latter being the most frequent cause of AKI.3 Sepsis presents with clinical manifestations of systemic inflammation combined with suspicion or evidence of an infection and is a major cause of multiple organ dysfunction and death in the ICU.4 The pathogenesis of sepsis-associated AKI (SA-AKI) is believed to be an interplay of systemic and renal inflammation, hypoxia and dysregulated renal bioenergetics.5 There are no pharmacological interventions approved for the treatment of SA-AKI. Currently, the only supportive treatment option available is renal replacement therapy (RRT).
A potent new candidate drug in clinical development to treat or prevent SA-AKI is alkaline phosphatase (AP). AP is a dephosphorylating, membrane-bound, endogenously occurring enzyme, exerting detoxifying effects through dephosphorylation of endotoxins, involved in sepsis pathogenesis6 ,7 and other proinflammatory compounds, including extracellular ATP.8 Several isoenzymes exist, namely intestinal, placental, germ-cell and tissue non-specific (liver/kidney/bone) AP, illustrating the broad expression throughout the body.9
Previously, clinical trials in healthy volunteers and patients with sepsis, with or without AKI, have established the tolerability and potential efficacy of purified bovine intestinal AP (biAP).10–12 In patients with SA-AKI, biAP significantly improved renal function according to the combined end point of endogenous creatinine clearance, requirement for RRT and duration of RRT. Moreover, a range of markers of systemic inflammation, renal function and renal damage in blood and urine demonstrated improvement, suggesting that a systemic anti-inflammatory effect induced by biAP prevented further renal injury. Following these encouraging results, a human recombinant AP (recAP) has been developed as a pharmaceutically acceptable replacement for bovine-derived AP. In order to improve enzyme stability, while maintaining catalytic function, the crown domain of a human intestinal AP is replaced with the crown domain of human placental AP.13 In line with preclinical and clinical studies using purified biAP, animal studies with recAP revealed potent anti-inflammatory activity preserving function14 and histological integrity of the affected kidneys (unpublished data) and administration to healthy volunteers did not raise any safety concerns.15 Therefore, therapeutic application of recAP is predicted to exert potent tissue protective and anti-inflammatory effects in patients with SA-AKI. In order to investigate this hypothesis, a randomised placebo-controlled phase II trial is being conducted with recAP in critically ill patients suffering from SA-AKI.
Methods and analysis
Design and setting
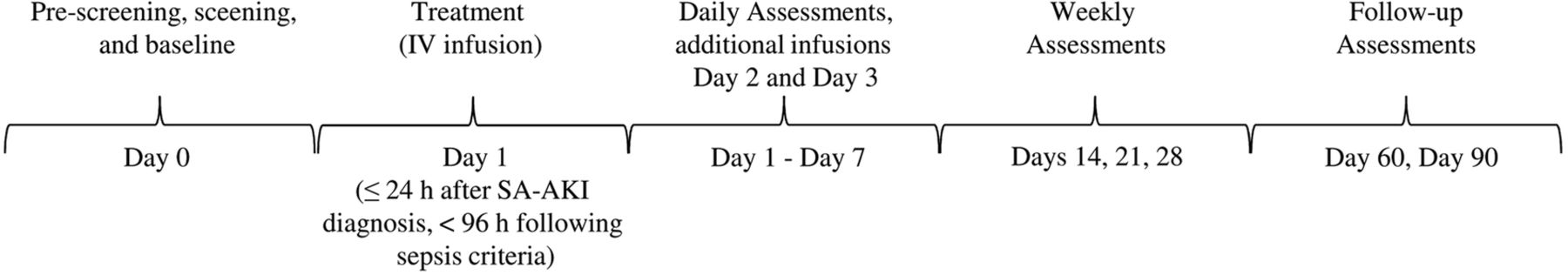
A randomised, double-blind, placebo-controlled, four-arm, parallel-group, proof-of-concept, dose-finding adaptive phase IIa/IIb study is being conducted in critically ill patients with SA-AKI. It is expected that the patients will be recruited by ∼50 sites across the European Union and North America. Participating institutions will be required to adhere to the Surviving Sepsis Campaign 201216 and 2012 KDIGO (Kidney Disease: Improving Global Outcomes) Clinical Practice Guideline for AKI recommendations.17 The study involves two parts with an interim analysis between the parts (figure 1), with continued recruitment during this interim analysis. Patients enrolled during Part 1 and during the interim analysis will be randomly assigned to receive either placebo or one of three different doses of recAP once daily for 3 days. The interim analysis on safety and the primary end point will be performed after the 120th patient of Part 1 has completed the day 7 visit of the study to select the dose to be administered in Part 2. The dose chosen will be the most efficacious dose of recAP on the primary end point in Part 1, provided there are no safety issues with that dose as judged by the data monitoring committee (DMC). In Part 2, patients will be randomly assigned to receive either placebo or the dose of recAP selected during the interim analysis. Patients recruited during the interim analysis period to the dose selected in Part 2 will form part of the Part 2 population, but those recruited to the doses that are not selected will be included in the Part 1 population. The duration of the study is defined for each patient as the date signed written informed consent is provided through the last follow-up visit on day 90 (figure 2). This study is reported in accordance with the Standard Protocol Items: Recommendations for Interventional Trial (SPIRIT) guidelines.18

Trial flow. ICF, informed consent form; recAP, human recombinant alkaline phosphatase; RRT, renal replacement therapy; SA-AKI, sepsis-associated acute kidney injury; h, hours.
{kind=link}
{kind=link}
Timeline. IV, intravenous; SA-AKI, sepsis-associated acute kidney injury; h, hours.
Study objectives
Primary objectives
The primary objectives are to investigate the optimal therapeutic dose and effect of recAP on renal function and related clinical parameters in patients with SA-AKI.
Secondary objectives
The secondary objectives are to investigate the safety and tolerability, pharmacokinetics (in the first 120 patients from Part 1 only), immunogenic potential and the effect on quality of life of recAP in patients with SA-AKI.
Other objectives
The other objectives are to determine whether specific patient groups benefit most from recAP treatment and whether patient groups that are non-responders can be identified. The identification of such groups will be based on several baseline characteristics, including kidney function markers and biomarkers for tubular injury and systemic inflammation.
Patient selection
A minimum of 290 patients will be enrolled, at least 120 patients in Part 1 and at least 170 patients in Part 2. Potential patients who have been admitted to the ICU will undergo a prescreening and, once informed consent is obtained, will undergo screening assessments to determine their eligibility. As soon as possible when all of the inclusion criteria and none of the exclusion criteria are met (box 1), and after confirmation of continuing AKI by either a fluid-corrected serum creatinine assessment or persistence of attenuated urine output, patients will be randomly assigned to a treatment group, undergo baseline determinations and start treatment with study drug (day 1). Treatment must be administered within 24 hours, at the latest, after SA-AKI is first diagnosed and within 96 hours from fulfilling sepsis criteria. Coincident with the interim analysis, but independent from it, some eligibility criteria (eg, age, weight, time-window of sepsis diagnosis, time-window of AKI diagnosis) were slightly modified to facilitate patient inclusion in the second part of the trial (see online supplementary table S1). These adjustments were considered to be unlikely to materially influence the characteristics of the subject population or to impact the potential risk–benefit of recAP. In addition, to achieve adequate participant enrolment, best practices will be shared via national coordinator meetings and if necessary, additional sites will be recruited and dormant sites will be closed.
Supplementary table
Patient entry selection
Inclusion criteria
Has informed consent form signed by patient or legal representatives or independent investigator, according to local rules and regulations
Is aged 18–85 years, inclusive
Is admitted to the intensive care unit or intermediate care unit
Has diagnosis of sepsis (<96 hours prior to first study drug administration or <72-hour prior to acute kidney injury (AKI) diagnosis), according to criteria defined by the American College of Chest Physicians/Society of Critical Care Medicine,19 based on:
A. A proven or strongly suspected bacterial infection
B. At least two of the following four SIRS criteria within a timeframe of 72 hours at the time of AKI diagnosis. Note: it is not required that symptoms are present simultaneously at study randomisation:
1. Core temperature >38°C or <36°C
2. Heart rate >90 bpm (unless the patient has a medical condition known to increase heart rate or is receiving treatment to prevent tachycardia
3. Respiratory rate >20 breaths/min, PaCO2 <32 mm Hg or the use of mechanical ventilation for an acute respiratory process
4. White cell count >12 000/mm3 or <4000/mm3 or a differential count showing >10% immature neutrophils
Has first diagnosis of AKI, defined as AKI Stage 1 or greater, according to the following AKIN criteria (Note: adjusted in regard to time-window):
A. Urinary output <0.5 mL/kg/h for >6 hours following adequate fluid resuscitation when applicable, in the absence of underlying primary renal disease, or
B. Increase (absolute) in serum creatinine >26.2 µmol/L (0.30 mg/dL) compared with a serum creatinine value within the previous 48 hours, or presumed to have occurred in the previous 48 hours when compared with a reference creatinine value (see below), or
C. Increase (relative) in serum creatinine to >150% (>1.5-fold) compared with a serum creatinine value in the previous 48 hours or presumed to have occurred in the previous 48 hours, when compared with a reference creatinine value (in the absence of primary underlying renal disease).
D. The reference creatinine value is a serum creatinine value in the following order of preference:
1. Lowest value within 3 months of the hospital admission. If not available:
2. At hospital admission. If not available:
3. At ICU admission. If not available:
4. Lowest value between 3 and 12 months prior to hospital admission
When the diagnosis of AKI is made according to one of the AKIN serum creatinine criteria, continuing AKI needs to be confirmed by a confirmative fluid-corrected serum creatinine measure, defined as no decrease in serum creatinine ≥26.2 µmol/L (≥0.30 mg/dL). The result must be available prior to randomisation. Administration of study treatment must be started within 24 hours after first AKI diagnosis
When the AKI diagnosis was made according to the AKIN urine output criteria, the oliguria or anuria should still meet the AKIN urine output criteria prior to randomisation. Administration of study treatment must be started within 24 hours after first AKI diagnosis
Exclusion criteria
Woman of childbearing potential with a positive pregnancy test (blood or urine), pregnant or breast feeding
Weighs more than 115 kg (253 lb)
Has life support limitations (eg, do not intubate, do not dialyse, do not resuscitate)
Is known to be HIV-positive
Has urosepsis*
Is already on renal replacement therapy (RRT) or a decision has been made to initiate RRT within 24 hours after planned start of study drug administration
Is receiving immunosuppressant treatment or is on chronic high doses (high-dose therapy exceeding 2 weeks of treatment) of steroids equivalent to prednisone/prednisolone 0.5 mg/kg/day, including solid organ transplant patients. Patients with septic shock treated with hydrocortisone (eg, 3×100 mg) can be included
Is expected to have rapidly fatal outcome (within 24 hours)
Has known, confirmed fungal sepsis
Has advanced chronic liver disease, confirmed by a Child-Pugh score of 10–15 (Class C)
Has acute pancreatitis with no established source of infection
Has participated in another investigational study within 30 days prior to enrolment into the study
Is not expected to survive for 28 days due to medical conditions other than SA-AKI, including cancer (previous haematological malignancies that are not actively treated allowable), end-stage cardiac disease, cardiac arrest requiring cardiopulmonary resuscitation or with pulseless electrical activity or asystole within the past 30 days, end-stage lung disease and end-stage liver disease
Has known prior history of CKD with a documented eGFR <60 mL/min by a commonly used formula such as MDRD or CKD-EPI, known GFR <60 mL/min or a known history of persistent creatinine level ≥150 μmol/L (1.70 mg/dL) prior to entry for reasons other than the current sepsis condition†
Has diagnosis of malaria or other parasite infections
Has burns on >20% of body surface
Has had AKI diagnosis according to the AKI inclusion criteria for a period longer than 24 hours prior to study drug administration
Is anticipated to be treated with non-continuous RRT from day 1 to day 7
During day 1 to day 7, continuous RRT is anticipated to be started or stopped not according to per protocol criteria
The AKI is most likely attributable to other causes than sepsis, such as nephrotoxic drugs (NSAIDs, contrast, aminoglycosides) and renal perfusion-related (acute abdominal aortic aneurysm, dissection, renal artery stenosis)
Improvement in serum creatinine of at least 26.2 µmol/L (0.30 mg/dL) prior to administration of the study drug
Patients who use nephrotoxic medication and who fulfil the SA-AKI inclusion criteria at screening are not eligible if the use of this nephrotoxic medication is planned to continue (eg, NSAIDs, ACE inhibitors, gentamycin, tobramycin)
Has a history of known intravenous drug abuse
Is an employee or family member of the investigator or study site personnel
Has active haematological malignancy
*As urogenital obstruction, frequently observed during urosepsis, may also account for an increase in serum creatinine levels, it is not possible to define which proportion might be due to SA-AKI. Therefore, urosepsis patients are excluded.
†CKD is a disease with a distinct pathophysiology compared with AKI. Also, if eGFR is impaired, the chances of an intervention to prevent further deterioration are limited. To increase homogeneity of the study population, these patients are therefore excluded.
AKI, acute kidney injury; AKIN, acute kidney injury network; CDK, chronic kidney disease; CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration; eGFR, estimated glomerular filtration rate; ICU, intensive care unit; IV, intravenous; MDRD, Modification of Diet in Renal Disease; NSAIDs, non-steroidal anti-inflammatory drugs; RRT, renal replacement therapy; SIRS, systemic inflammatory response syndrome.
Randomisation
Patients will be randomly assigned to receive either placebo or one of three doses of recAP using a 1:1:1:1 allocation ratio. The randomisation schedule will be stratified by site. An independent statistician will generate a permuted block randomisation schedule for an interactive voice/web response system (IVRS/IWRS), which will link sequential patient randomisation numbers to treatment codes. After informed consent has been obtained, the IVRS/IWRS provides a patient number at screening and retains a unique randomisation number for all patients. Once the screening assessments are performed, data have been reviewed by an investigator and the patient is considered to be eligible, the investigator will contact the IVRS/IWRS to randomly assign the patient a treatment number through IVRS/IWRS. Once it has been decided which is the most efficacious dose of recAP on the primary end point to be administered in Part 2, the codes for the treatment groups corresponding to the two dropped doses will be discontinued, and 1:1 treatment allocation will continue using the codes for the remaining treatments (1 active, 1 placebo).
Informed consent
Informed consent (see online supplementary appendix I) will be obtained before any study-specific procedures are performed. Country-specific regulations regarding the use and collection of personal data and biological specimens, ancillary care, and financial care to those who suffer from harm will be followed at each site. It is anticipated by the very nature of the study that many patients who will be eligible for this protocol due to their severe underlying condition will not be able to provide informed consent themselves for various reasons. In a situation where a patient is unable to provide consent, the patient's legally authorised representative may provide written consent as approved by the institutional-specific guidelines. Informed consent may be obtained from an independent consulting physician in countries where regulation and institution guidelines permit. This form of consent can only be used when the patient and legally authorised representative are unable to provide consent. In cases where the initial informed consent is obtained from a legal representative or an independent consulting physician, the patient also must consent as soon as they are able. The investigator shall retain the signed original Informed Consent Form (ICF) and give a copy of the signed original form to the patient or legal representative.
Supplementary appendix
Blinding
All people involved in the study will be blinded to treatment assignment. The randomisation schedule will be held by an independent, non-blinded statistics team at a different regional location and will not be revealed until all patients have completed the study and the database has been finalised for the end of the study. The non-blinded interim analysis will be conducted and delivered to the independent data monitoring committee by the non-blinded biostatistics team, located at a different site to the blinded team. A patient's treatment assignment will not be broken until the end of the study, unless knowledge of the treatment assigned to the patient is necessary to guide medical treatment of the patient. The determination of AP activity in plasma, often part of the routine clinical chemistry panel, could lead to non-blinding and to erroneous interpretation of liver function, as the recAP administered will increase the AP activity, exceeding many times the reference range AP levels. Therefore, AP activity levels from samples taken during the first 14 days of the study are not allowed to be determined/reported to the study team members or to any other study staff member involved in the conduct of the study.
Study intervention
Patients randomly assigned to receive recAP in Part 1 will receive one of the following three doses of recAP: 0.4 mg/kg (250 U/kg), 0.8 mg/kg (500 U/kg) or 1.6 mg/kg (1000 U/kg). Study drug will be administered by 1-hour intravenous infusion as soon as possible, but within 24 hours after SA-AKI is first diagnosed, on day 1, and 24±1 hours later on days 2 and 3. A total volume of 50 mL will be infused at a constant rate of 50 mL/hour. The preferred route for study drug administration is through a central venous line; if this is not possible, a peripheral line is acceptable. Study drug is being administered separately from any concomitant drugs using a dedicated lumen of the catheter. At the start of each drug administration, the exact volume of recAP or placebo to be administered to each patient will be calculated on the basis of the patient's hospital admission weight. Patients weighing between 95 and 115 kg will receive the same dose as that for patients weighing 100 kg. The volume of the placebo and the volume of the active doses of recAP are identical. RecAP is supplied as a clear, colourless, pyrogen-free solution for intravenous infusion with a content of 40 mg per phial at 8.0 mg/mL in an aqueous buffer. Matching placebo is supplied in an identical manner as the same aqueous buffer. RecAP and matching placebo will be prepared in randomised treatment packs comprising three daily doses and shipped to the clinical site. Each pack will contain a (randomised) dosage for one patient and will contain a sufficient quantity for dispensing during the 3-day double-blind treatment period. The content of the vials will be used to fill an intravenous dosing syringe and will be dose adjusted to an appropriate volume corresponding to the body weight of the patient, followed by the addition of physiological saline solution to a total of 50 mL. All study drug is to be stored in a lockable storage facility under appropriate and monitored pharmacy conditions.
Concomitant medications
After randomisation, the administration of nephrotoxic drugs, such as contrast agents, non-steroidal anti-inflammatory drugs, ACE inhibitors, angiotensin receptor blockers, gentamycin or tobramycin should be avoided where possible, as recommended in the KDIGO Clinical Practice Guideline for AKI recommendations.17
Outcome measures
Primary end point
The primary end point is the area under the time-corrected endogenous creatinine clearance curve from day 1 (first measurement after treatment) to day 7, inclusive (AUC1–7), with the creatinine clearance curve being the mean creatinine clearance on each day. Time-corrected endogenous creatinine clearance is assessed on each day during a 6±1 hour period with serum creatinine measurements before and after the urine collection period to calculate in mL/min as the mean creatinine clearance over the period. Although 24-hour urine collection periods are the most accurate to reliably determine creatinine clearance, a 6-hour period is expected to be representative of the full 24 hours for that day.20 ,21 If there is no diuresis, the clearance will be reported as 0 mL/min.
AUC1–7 is calculated as the average of the time-corrected endogenous creatinine clearance values over the 7 days. Specifically, denoting Ci as the mean time-corrected endogenous creatinine clearance on day i, AUC1–7 is defined as:
 Any missing Ci values will be handled by linear interpolation where possible, otherwise they will be imputed by last observation carried forward. When there are no preceding postbaseline measurements to use, the baseline measurement from day 0 (prior to treatment) will be carried forward.
Any missing Ci values will be handled by linear interpolation where possible, otherwise they will be imputed by last observation carried forward. When there are no preceding postbaseline measurements to use, the baseline measurement from day 0 (prior to treatment) will be carried forward.
Key secondary end point
The key secondary end point is RRT incidence during the period day 1 (after first treatment) to day 28, inclusive. Should a patient die or withdraw from the study during this period without recording RRT incidence, he or she will be counted as having not required RRT. From day 1 to day 7, only continuous modalities of RRT are allowed. From day 8 onward, intermittent/non-continuous modalities such as intermittent haemodialysis are allowed. Criteria for the initiation and timing of stopping of RRT are described in box 2. Although these criteria are strongly preferred within the protocol setting, based on clinical judgement, investigators may deviate from these criteria.
Start and stop criteria RRT
Initiation of RRT22*
Anuria (negligible urine output for 6 hours)
Severe oligura (urine output <200 mL over 12 hours)
Hyperkalaemia (potassium concentrations >6.5 mmol/L)
Severe metabolic acidosis (pH <7.2 despite normal or low partial pressure of carbon dioxide in arterial blood)
Volume overload (especially pulmonary oedema unresponsive to diuretics)
Pronounced azotaemia (urea concentrations >30 mml/L or creatinine concentrations >300 μmol/L)
Clinical complications of uraemia (eg, encephalopathy, pericarditis, neuropathy)
Timing of stopping RRT23
If (on continuous RRT or between intermittent haemodialysis sessions) diuresis >30 mL/hour and there are no other indications for RRT, then endogenous creatinine clearance should be calculated using a 6-hour urine collection period:
A. If endogenous creatinine clearance ≥20 mL/min, RRT should be discontinued
B. If endogenous creatinine clearance ≤12 mL/min, RRT should be continued
C. If endogenous creatinine clearance >12 and <20 mL/min, continuation or termination will be the decision of the treating physician
*Meeting one criterion makes the patient eligible for initiation of RRT.
RRT, renal replacement therapy.
Other secondary end points
Other secondary renal end points include the duration of RRT over 28 days and renal function at day 28 (creatinine clearance if available, or estimated GFR (eGFR) according to the chronic kidney disease epidemiology collaboration (CKD-EPI) formula) and at days 60 and 90 (eGFR). Clinical end points include liver enzymes (aspartate aminotransferase, alanine aminotransferase, γ-glutamyl transpeptidase, lactate dehydrogenase, bilirubin), lung function (PaO2/FiO2 ratio, positive end-expiratory pressure and tidal volume), mechanical ventilation over 28 days, shock over 28 days, SOFA scores and mortality over 90 days. Biomarker end points include kidney function markers (urine and serum creatinine, blood urea nitrogen, sodium, proteinuria, fractional excretion of sodium and urea and urine volume), biomarkers for tubular injury (kidney injury molecule-1, interleukin (IL)-18 and α-glutathione s-transferase) and systemic inflammation (IL-6, C reactive protein and lipopolysaccharide (LPS)-binding protein), and recAP pharmacokinetic properties.
Additional end points include composite end points (combinations of need for RRT, mortality, eGFR and hospitalisation for new AKI episode), serology, assessed by IgG, IgE and total immunoglobulin, quality of life, assessed by the EuroQol-5D questionnaire and ICU and hospital length of stay over 90 days. The safety analyses include (serious) adverse events (SAEs), laboratory assessments (eg, antidrug antibodies), haematology, vital signs and electrocardiography data.
Statistical and analytical plan
Sample size calculations
A sample size of n1=30 patients per treatment group in Part 1 with an additional n2=85 patients recruited to the optimal recAP dose and placebo treatment groups in Part 2 (for a total sample size of n=290 patients) is planned. Custom-programmed simulations were performed using SAS software V.9.2 to determine power and type I error rate of the chosen sample size and design under a number of different dose–response scenarios. Each scenario assumed an SD of 49 mL/min for the primary end point with an assumed response of 60 mL/min for the placebo group, and between 60 mL/min (no treatment effect) and 79 mL/min (strong treatment effect) for the recAP dose groups.
Fifty thousand simulations were performed to show that the one-sided type I error rate is 2.4% (and hence is well controlled at the one-sided 2.5% significance level). The power was defined as the probability of rejecting the null hypothesis (of no difference between treatment groups) when one or more recAP dose groups have an effective treatment effect, defined as a response of 69.5 mL/min. This was investigated across 7 scenarios with 10 000 simulations performed for each. The chosen design achieved power of between 79% and 86% for scenarios with strong treatment effects for the medium and high recAP dose groups and a varying response for the low-dose group, and between 66% and 67% when only the high-dose group had a strong treatment effect.
As the sample size determination is based on the number of patients required for the intention-to-treat (ITT) analysis, patients who are randomly assigned and subsequently withdraw prior to completion of the study will not be replaced.
Interim analysis
A non-blinded interim analysis will be conducted on the Part 1 data to determine the optimal recAP dose for Part 2. This analysis will compare the three recAP doses and placebo from Part 1 on the primary efficacy end point and the safety data. The optimal dose is defined as the dose with the best safe improvement on creatinine clearance compared with placebo. Unless the DMC have safety concerns pertaining to specific dose level(s) of recAP and/or the dose–response modelling of the primary end point (Emax model) highlights unusual trend(s), the optimal dose will be, preferably, the highest dose of recAP. If there are safety concerns and/or concerns with regard to the dose–response curve, the optimal dose level will be selected among the dose levels where there are no safety or/and dose–response modelling concerns. In that case, the effectiveness of a dose level will be determined based on the one-sided unadjusted p value for comparison with placebo on the primary end point, that is, the dose with the smallest p value will be considered the most effective dose. Should more than one dose appear to be the most effective with the same effect, the safety data as well as the dose–response modelling should be closely examined for selecting the optimal dose.
Safety data being analysed include the use of concomitant medications, adverse events (AEs), treatment emergent AEs (TEAEs) and SAEs, laboratory assessments and vital signs.
The results will only be reviewed by an independent non-blinded DMC, who will make the dose selection decision. The selected dose will be reported to the sponsor in a blinded fashion. The study will not be prematurely terminated in case a statistically significant benefit is found during the interim analysis.
The study may be terminated for futility if none of the three recAP doses in Part 1 show evidence of efficacy for the primary end point. Each recAP dose will only be deemed futile if the one-sided, unadjusted p value for its comparison with placebo is <0.8. If all of the doses in Part 1 fulfil the futility criterion, then the DMC will recommend further discussion with the sponsor and Steering Committee to determine whether the trial should be terminated for futility.
Statistical analyses
Statistical analyses will be performed using SAS software V.9.2 or later. Continuous variables will be summarised using descriptive statistics, and categorical variables will be summarised using frequency counts and percentages. All statistical tests, primarily performed on the ITT set, will be two-sided and performed using a 5% significance level, leading to 95% (two-sided) CIs.
The analysis of the primary efficacy end point will be performed separately for Parts 1 and 2: for Part 1, AUC1–7 will be compared between the three recAP doses and placebo by an analysis of variance (ANOVA) of the AUC1–7 with treatment and site as explanatory variables, or corresponding non-parametric analyses if appropriate. For Part 2, the optimal recAP dose will be compared with placebo using the same ANOVA model or corresponding non-parametric analyses if appropriate. To address multiplicity, the type 1 error rate will be controlled by combining the results for the primary efficacy end point from the interim analysis (Part 1) and the final analysis (Part 2), using the inverse normal method.24
A hierarchical method will be employed to address any multiplicity arising from the analysis of the key secondary end point, indicating that the optimal recAP dose will be formally compared with placebo, using a logistic regression model with treatment group and site as explanatory variables, only if a statistically significant result is obtained from the combination test analysis of the primary end point. Otherwise, the results will be reported as exploratory analysis only.
All analyses performed on the other secondary end points are for exploratory purposes only. Therefore, no further multiplicity adjustment is required.
Data quality assurance
The sites will maintain source documentation and enter patient data into the electronic case report form (eCRF) as accurately as possible. eCRFs are accessed through Medidata Rave (Medidata Solutions, New York, New York, USA). This electronic data capture system is validated and compliant with US Title 21 Code of Federal Regulations (CFR) Part 11. Each person involved with the study will have an individual user name and password that allows for record traceability. A quality review of the data will be performed by the site with additional reviews by the clinical monitor through 100% source data verification. Each eCRF is presented as an electronic copy, allowing data entry by study site staff, who can add and edit data, identify and resolve discrepancies, and view records. This system provides immediate direct data transfer to the database, as well as immediate detection of discrepancies, enabling site coordinators to resolve and manage discrepancies in a timely manner. After all data reviews and query resolutions are complete, the statistical analysis plan approved and signed, and any summary/analysis populations approved, the database will be locked.
Safety assessment
Medication errors
Since the study drug will be prepared, double-checked and administered by trained hospital staff, and since the dose levels under study have not caused any SAEs in healthy volunteers,15 the risks for potential hazardous or life-threatening events due to medication errors are considered to be minimal.
Overdose
In healthy volunteers, the maximum administered single dose was 3.2 mg/kg (2000 U/kg) and this dose did not result in any SAEs.15 As an example of a possible dose error (eg, administration of the highest study dose for a 100 kg person to a 50 kg person), 1-day administration of study drug still would not exceed the tested maximum dose in healthy volunteers.
AEs report
All patients will be monitored closely for signs of adverse reactions. AEs will be reported in the eCRF from the time the patient signs the ICF until day 28 visit for all AEs (including SAEs) regardless of its relationship to study drug. Any AE that occurs during or after the first dose of study drug is considered treatment emergent. The intensity of the AE will be rated as mild, moderate or severe. In addition to patient observations, TEAEs will be documented from any data collected on the AE page of the eCRF (eg, laboratory values, physical examination findings) or identified from review of other documents (eg, patient diaries) that are relevant to patient safety. When patients have left the hospital, they will be instructed to contact the investigator at any time if any symptoms develop. In the event of an SAE, the investigator may immediately stop treatment if it is considered in the best interest of the patient. In case study drug is discontinued early, the patient will continue follow-up in the study as per protocol. The assessment of the relationship of an AE to the administration of study drug (unrelated, possible, probable, definite) is a clinical decision based on all available information at the time of completion of the eCRF.
External data monitoring committee
An independent DMC will review safety and efficacy data during Part 1 and Part 2 of the study at predefined milestones. The DMC consist of three members with experience in independent clinical review boards: two clinical experts and one biostatistical expert. The DMC is supported by two clinical pharmacology advisors.
Withdrawal
Patients have the right to withdraw from the study at any time and for any reason. Investigators should attempt to determine the cause of withdrawal and, if agreed by the patient, let the patient return for the day 90 visit. The extent of a patient's withdrawal from the study (ie, withdrawal from further study treatment, withdrawal from active participation in the study, withdrawal from any further contact) should be documented. Treatment with study drug will be stopped when continued treatment with study drug is not in the best interest of the patient, if the patient withdraws consent, when investigator or sponsor decides to discontinue the patient's participation in the study or when AM-Pharma terminated the study. Additionally, treatment with study drug may be stopped in case of protocol non-compliance or deviations. In case study drug is discontinued early, the patient will continue follow-up in the study as per protocol to allow for ITT analysis.
Study period
Preparations of the study are completed. The study started enrolling patients in December 2014. Part 1 is completed and the most efficacious dose of recAP has been selected during the interim analysis in April 2016. The estimated study enrolment completion date of Part 2 is February 2017. Of note, this manuscript was completed prior to the interim analysis.
Ethics and dissemination
Ethics
Before study onset, the protocol, amendments, informed consent and any other written information regarding this study to be provided to the patient or the patient's legal guardian were approved by the relevant institutional review boards (IRBs)/independent ethics committees (IECs). Amendments will be communicated to all relevant parties via mail, investigator meetings and site trainings. The study is being performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki, ICH E6(R1), Code of Federal Regulations and all other applicable regulations. Fully informed consent will be obtained before any study-specific procedures are performed. Country-specific regulations regarding the collection of personal data will be followed at each site.
Potential conflicts of interest
During this trial, AM-Pharma, the manufacturer of recAP, has no involvement in the randomisation, interim analyses and dose selection, and analyses and interpretation of data. AM-Pharma is involved in drafting of the manuscript.
Confidentiality
All laboratory specimens, evaluation forms, reports and other records will be identified in a manner designed to maintain patient confidentiality. All records will be kept in a secure storage area with limited access. Clinical information will not be released without the written permission of the patient (or the patient's legal guardian), except as necessary for monitoring and auditing by the sponsor, its designee, the Food and Drug Administration, the European Medicines Agency, other regulatory national authorities or the IRB/IEC. The investigator and all employees and coworkers involved with this study may not disclose or use for any purpose other than performance of the study any data, record or other unpublished, confidential information disclosed to those individuals for the purpose of the study. Prior written agreement from the sponsor or its designee must be obtained for the disclosure of any said confidential information to other parties.
Monitoring of the study
The clinical monitor, as a representative of the sponsor, has the obligation to follow the study closely. In doing so, the monitor will visit the investigator and study site at periodic intervals, in addition to maintaining necessary phone and letter contact. The monitor will maintain current personal knowledge of the study through observation, review of study records and source documentation, and discussion of the conduct of the study with the investigator and personnel.
Dissemination policy
After completion of the study, the data will be reported at scientific meetings and published in a peer-reviewed scientific journal, regardless of the outcome. Authorship will be based on the criteria of the International committee of medical journal editors (ICMJE). There is no intention to use a professional writer.
Discussion
RecAP is one of the limited pharmaceutical treatment options for SA-AKI currently being tested in a clinical trial setting. Despite several attempts, many promising new treatment strategies for AKI have failed when tested in patients.25–27 Potential explanations include the heterogeneity of the study population and the complex pathogenesis of this disease.5 Of interest, bovine-derived intestinal AP has shown to improve renal function when administered to critically ill patients with SA-AKI.11 ,12 To further improve the renal-protective potential of AP in the treatment of SA-AKI, recAP was developed by replacing the crown domain of a human intestinal AP with the crown domain of human placental AP.13 This results in a more enzymatically stable, yet catalytically active enzyme, as confirmed by the phase I trial in healthy volunteers.15 To investigate the efficacy of recAP in the treatment of critically ill patients with SA-AKI, we are conducting a large multicentre phase II clinical trial.
It is thought that recAP exerts a dual mechanism of action, targeting this multifactorial pathogenic response. First, recAP dephosphorylates and thereby detoxifies LPS,6 ,7 a part of Gram-negative bacteria involved in the development of sepsis, which can elicit an inflammatory response systemically and locally within the kidney on binding to toll-like receptor 4.28 ,29 Second, recAP dephosphorylates extracellular ATP into the cytoprotective and anti-inflammatory signalling molecule adenosine.14 Since recAP mimics the activity of an endogenous enzyme and considering the dual mechanism of action, recAP might be superior to other agents tested in the past as already suggested by the previous clinical trials performed with biAP.11 ,12
In this study, sepsis is defined based on a proven or strongly suspected bacterial infection, and the presence of at least two of the four SIRS criteria (box 1). However, the validity of SIRS criteria to define sepsis and the previously recognised key role of immune-modulating mechanisms in sepsis pathogenesis are currently under debate. As such, new definitions of sepsis and septic shock have been recently proposed, which states that sepsis is a life-threatening organ dysfunction caused by a dysregulated host response, and defines sepsis as a suspected or documented infection and the presence of organ dysfunction, defined as an acute increase of ≥2 SOFA points for one organ system.30 As our study was designed preceding publication of these new criteria, implementation of these criteria was not feasible. However, we expect that we overcome the drawbacks of the original criteria, as this study only includes patients with sepsis combined with a failing organ system, namely the kidney.
As there are no guidelines for the development of drugs for SA-AKI, the study was designed by a group of leading global experts in AKI and sepsis, with input from the US Food and Drug Administration and the European Medicines Agency and several local European country authorities. The resulting design was a dose-finding adaptive trial. The heterogeneity of the study population, strict inclusion criteria and narrow time-windows may limit generalisation to the entire population of critically ill patients with SA-AKI. On the other hand, the occurrence of non-responders, or specific patient groups benefitting the most of recAP treatment will be investigated, which may also provide information about the potential of recAP treatment for other nephropathies as well.
Results of this randomised controlled phase II study will be of importance for critically ill patients with SA-AKI, as they will allow us to draw conclusions on the efficacy of recAP in the improvement of renal function and related clinical parameters.
Acknowledgments
The authors are grateful to all national coordinators for their contribution. This includes Martin Matejovic (Czech Republic), Jacques Creteur (Belgium), Ville Pettilä (Finland), Didier Payen de la Garanderie (France), Antonio Artigas (Spain) and Mervyn Singer (UK).
References
Footnotes
JA and PP share senior authorship.
Collaborators RLM, PTM, JH, MJ, PP.
Contributors EP and PP drafted the manuscript. All authors participated in the conception, design and/or coordination of the study. JA, RLM, PTM, JH, MJ and JAK helped revising the manuscript. All authors read and approved the final manuscript for publication.
Funding This work was funded by AM-Pharma.
Competing interests JAK, MJ, RLM and PTM have received consultancy fees for the design of this trial from AM-Pharma. PP has received travel reimbursements, and consultancy fees for the design of this trial from AM-Pharma. JA has a patent pending and is an employee of AM-Pharma. JH is an employee of the CRO sponsored to conduct this study. EP declares to have no relevant financial interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The full trial protocol can be accessed by contacting PP. The list of study sites can be obtained by contacting JA and is published on clinicaltrials.gov.