Article Text

Abstract
Introduction The invasive pneumococcal diseases (IPDs) caused by Streptococcus pneumoniae pose an enormous threat to children under 5 years of age. However, routine use of pneumococcal conjugate vaccines could aid in reducing the incidence of IPDs. The purpose of this clinical trial is to assess the non-inferiority of the investigational 13-valent pneumococcal conjugate vaccine (PCV13) to the currently licensed 7-valent pneumococcal conjugate vaccine (PCV7).
Methods and analysis 1040 infants will receive a three-dose series of either PCV13 or PCV7 at ages 3, 4 and 5 months, respectively, and a booster dose at 12–15 months. Primary end points are the percentage of participants reaching a serotype-specific IgG concentration of ≥0.35 µg/mL and the IgG antibody geometric mean concentrations (GMCs) measured 30 days after the primary immunisation. Secondary end points include the percentage of vaccine recipients reaching a serotype-specific IgG concentration threshold of 1.0 µg/mL, the percentage of participants reaching the pneumococcal opsonophagocytic assay (OPA) titre threshold of 1:8, and the geometric mean titres (GMTs) of OPA measured 30 days after primary and booster doses. The number of standard IgG responders and IgG GMCs measured 30 days after the booster immunisation will also be determined. To evaluate differences between two groups, the sequential testing of the non-inferiority of PCV13 for the seven common serotypes and its effectiveness in treating the six additional serotypes will be performed.
Ethics and dissemination Ethics approvals have been granted by the Ethics Committees at the three provinces involved in this study: Shanxi, Henan and Hebei. The trial will be reported in accordance with the CONSORT guidance.
Trial registration number NCT02736240.
- Pneumococcal conjugate vaccine
- Sequential test
- Non-inferiority
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first study to investigate the immune responses induced by a locally developed and manufactured 13-valent pneumococcal conjugate vaccine (PCV13) among Chinese infants.
A total of 1040 infants will be recruited to support the randomised non-inferiority study design and a sequential test method is adopted.
Primary outcomes are proportions of vaccine recipients achieving a predefined specific-serotype IgG concentration threshold and geometric mean IgG antibody concentrations measured 30 days after primary immunisation, both of which are recommended by the WHO.
Besides the complete evaluation of immunogenicity and safety of the new PCV13, not involving the assessment of concomitant immunisation with other vaccines specified in paediatric vaccination might be seen as a potential limitation of this trial.
Introduction
Background
Streptococcus pneumoniae (S. pneumoniae or pneumococcus) is a leading cause of bacterial pneumonia, meningitis and sepsis in children, and thus poses a major threat to public health around the world. According to a WHO investigation in 2005, an estimated 0.7 to 1 million children under the age of 5 years die from pneumonia every year, most of whom lived in developing countries.1 The top 10 countries with the highest number of reported pneumococcal cases were all in Asia and Africa, with China accounting for 12% of the total.2 Immunisation against Hib, pneumococcus, measles and pertussis is the most effective way to prevent pneumonia.3 Routine use of the 7-valent pneumococcal conjugate vaccine (PCV7) has resulted in a dramatic reduction in the incidence of invasive pneumococcal disease (IPD) attributable to pneumococcal serotypes contained in the vaccine.4 However, there is still an urgent need for a pneumococcal conjugate vaccine with expanded coverage for IPD resulting from serotypes not included in PCV7.5
On 24 February 2010, the US Food and Drug Administration licensed a 13-valent pneumococcal conjugate vaccine (PCV13) for children from 2 months to 5 years of age. Since then, several studies have been conducted on PCV13 in many countries except China. The licensed PCV13 has been shown to be as effective as PCV7 in the prevention of disease caused by the seven common serotypes and could provide expanded protection against the six additional serotypes with a comparable safety profile.6 When coadministered with diphtheria, tetanus and acellular pertussis vaccine (DTaP), PCV13 is well tolerated and as immunogenic as PCV7.7 The flexible primary immunisation schedules of PCV13 for infants found no significant differences in antibody levels for almost all serotypes after the booster dose at 12 months of age8 ,9 and is thus recommended by the WHO.10 Furthermore, the clinical efficacy of PCV13 for multiple purposes in age groups ranging from 6 to over 70 years of age has been confirmed.11–16 Nevertheless, it is still unknown whether the licensed PCV13 or a new PCV13 will be effective for the Chinese population until now.
To address the issue, we plan to initiate this trial for a prelicensing evaluation of a new PCV13. The investigational PCV13 (batch number 201511003) is manufactured by Yuxi Walvax Biotechnology Co (an affiliate of Walvax Biotechnology Co) and supplied in penicillin bottles. Each 0.5 mL dose contains 2.2 µg pneumococcal polysaccharide serotypes 1, 3, 4, 5, 6A, 7F, 9V, 14, 18C, 19A, 19F and 23F and 4.4 µg pneumococcal polysaccharide serotype 6B; each serotype is conjugated to a tetanus toxoid carrier protein and adsorbed on aluminium phosphate. In accordance with the guidelines formulated by the WHO,17 and the requirements established by national regulatory authorities, licensing a new vaccine requires randomised clinical trials to show the non-inferiority of the new vaccine to an existing pneumococcal conjugate vaccine. PCV7 will serve as the control vaccine, since it is the only pneumococcal conjugate vaccine that has been licensed in mainland China. The control PCV7 (Prevnar, batch number H29315H13880) is manufactured by Wyeth and supplied in prefilled 0.5 mL syringes. Each 0.5 mL dose contains 2 µg pneumococcal polysaccharide serotypes 4, 9V, 14, 18C, 19F and 23F and 4 µg pneumococcal polysaccharide serotype 6B; each serotype is conjugated to a CRM197 carrier protein and adsorbed on 0.5 mg aluminium phosphate.
Objectives
The purpose of this study is to (1) determine if the seven common serotype-specific pneumococcal IgG responses elicited by PCV13 are all non-inferior to those elicited by PCV7 and (2) determine if the six additional serotype-specific pneumococcal IgG responses induced by PCV13 are all clinically effective 30 days after the primary immunisation.
We hypothesise that for each of the seven common pneumococcal serotypes (4, 6B, 9V, 14, 18C, 19F, 23F) (1) the lower bound of the 97.5% (98.75%, one-sided) CI for the difference (PCV13—PCV7) in the proportion of vaccine recipients reaching the serotype-specific IgG concentration threshold of 0.35 μg/mL is no < −10%; or (2) The lower bound of the 97.5% (98.75%, one-sided) CI of the serotype-specific IgG geometric mean concentration (GMC) ratio (PCV13/PCV7) is > 0.5 (−0.3, logarithmically transformed by base 10).
As a parallel hypothesis, we expect that for each of the six additional pneumococcal serotypes (1, 3, 5, 6A, 7F, 19A) (1) The proportion of the serotype-specific IgG responders achieving an IgG threshold of 0.35 μg/mL in the PCV13 group is greater than that of the PCV7 group; and (2) the lower limit of the 95% (2-sided) CI for the proportion of vaccine recipients reaching a serotype-specific IgG concentration threshold of 0.35 μg/mL is ≥70%.
Trial design
This study is a multicentre phase III randomised parallel group, allocation concealed non-inferiority trial with two groups to evaluate the immunogenicity and safety of PCV13 compared with PCV7 among healthy infants in China.
Methods
Study setting
The study includes six sites scattered in three provinces in North China. Each site has been ratified by the Center for Food and Drug Inspection, a department of China Food and Drug Administration, to join the research team of this trial and recruit participants.
Eligibility criteria
Inclusion criteria
Participants will only be recruited when he or she meets all of the following criteria at the time of randomisation:
Healthy infants, as established by medical history and clinical examination before entering into the study, who are 3 months old at the time of the first vaccination
Obtained written informed consent from the legally responsible representative(s)
Have the compliance of the participants' legally responsible representative(s) with the requirements of the protocol
No pneumococcal vaccine history or use of other preventive vaccine at least 7 days before study entry
Axillary temperature ≤37°C
In addition, participants must not encounter any of the following exclusion criteria:
Vaccinated by on sale or other on trial pneumococcal vaccines
Have had an invasive disease with S. pneumoniae as the verified cause
Severely allergic to any vaccines or drugs, or sensitive to temperatures ≥39°C associated with biological products inoculation
Birth weight <2.5 kg
History or family history of convulsion, seizure, encephalopathy or neurological disorder
Born from abnormal labours (difficult labours, aided by instruments) or have a history of asphyxia or nerve damage
Confirmed thrombocytopenia or a history of blood coagulation disorder
Confirmed pathological jaundice
Any confirmed or suspected immunodeficiency involving immunosuppressive therapy (radiotherapy, chemotherapy, corticosteroid hormone, antimetabolite, cytotoxic drugs), HIV infection, etc
Any confirmed or suspected congenital defect or serious chronic illnesses (trisomy 21 syndrome, diabetes, sickle cell anaemia, neurological disorder or Guillain-Barré syndrome)
Any confirmed or suspected diseases including respiratory diseases, acute infection, active period of chronic disease, cardiovascular disease, hepatic-nephrotic disease, cancer and dermatosis
Administration of immunoglobulin and/or any blood products, except hepatitis B hyperimmune globulin (HBlg)
Concurrent participation in another clinical study
Other exclusion criteria affirmed by investigators
Intervention
Eligible infants will be randomised to either the PCV13 or the PCV7 group to receive a three-dose series of PCV13 or PCV7 at ages 3, 4 and 5 months, respectively and a booster dose between 12 and 15 months. For all participants, blood samples will be obtained at four different time points: immediately before the first dose, 30 days after the primary series, immediately before the booster dose, and 30 days after the toddler dose.
Serum concentrations of anticapsular polysaccharide IgG for each of the 13 pneumococcal serotypes will be measured for all participants at the previously mentioned time points, using the standardised ELISA method.18 Additionally, serum functional opsonophagocytic activity (OPA) for each serotype will be assayed at the last 3 time points for a randomly selected subset of ∼100 participants in each group (200 infants in total), composed of infants assigned the first 20–36 participant numbers from each of the six sites.
Each participant will be observed for 30 min after each vaccination for any immediate reaction by the research staff. Any noted adverse events (AEs) will be recorded at that time. After leaving the site, local reactions (pain, redness, swelling and pruritus), systemic events (somnolence, vomiting, diarrhoea, crying and irritability), axillary temperature, other AEs and concomitant medications to treat or prevent symptoms will be recorded in daily cards by the parents/legally responsible representatives for 8 days. On the eighth day after vaccination, the documents will be returned to the site. AEs occurring between the 8th and the 30th day after each dose together with the usage of relevant concomitant medications will be recorded in connection cards and handed in at the next visit. Serious adverse events (SAEs) will be collected throughout the study until 6 months after the last dose. The intervention procedures are shown in table 1.
The intervention procedure
Outcome measures
Primary immunogenicity outcomes
For each of the 13 pneumococcal serotypes covered in PCV13:
Proportion of vaccine recipients reaching the serotype-specific IgG concentration threshold of 0.35 μg/mL 30 days after the primary series.
GMC of serotype-specific IgG antibodies 30 days after the primary series.
Secondary immunogenicity outcomes
For each of the 13 pneumococcal serotypes covered in PCV13:
Proportion of vaccine recipients reaching the OPA threshold of 1:8 30 days after the primary series.
Geometric mean titre (GMT) of OPA 30 days after the primary series.
Proportion of vaccine recipients reaching the serotype-specific IgG concentration threshold of 1.0 μg/mL 30 days after the primary series.
Proportion of vaccine recipients reaching the serotype-specific IgG concentration threshold of 0.35 μg/mL 30 days after the booster dose.
GMC of serotype-specific IgG antibodies 30 days after the booster dose.
Proportion of vaccine recipients reaching an OPA threshold of 1:8 30 days after the booster dose.
GMT of OPA 30 days after the booster dose.
Proportion of vaccine recipients reaching a serotype-specific IgG concentration threshold of 1.0 μg/mL 30 days after the booster dose.
Safety outcomes
Safety outcomes include all recorded local reactions and systemic AEs that occur within the 30 days following the primary series and booster doses and the percentage of vaccine recipients that reported each AE.
Trial duration
The study will last for 26 months. If unsuspected acute abnormal reactions occur during the trial, or the daily proportion of participants who experienced AEs rated grade 3 or above reaches 15% after any dose vaccination, the trial should be suspended. A panel will be involved in discussions of the safety profile and decide whether the study should be continued.
Sample size
For each of the seven common serotypes in PCV13 and PCV7: On the basis of data from previous studies, we assume that the percentage of vaccine recipients reaching the serotype-specific IgG concentration threshold of 0.35 μg/mL will be 85%.19 The non-inferior margin for the trial will be set at 10%. If the non-inferiority does exist for a proportion of the test group, a sample size of 434 infants per group, 868 total, will provide 80% power to ensure that the lower limit of 97.5% two-sided CI for difference (test group–control group) will be no < −10%. We also postulate that the common variance of serotype-specific IgG GMC, which has been logarithmically transformed by base 10, will not be >1.0. A sample size of 379 infants per group, 758 total, will have 80% power to detect whether the lower limit of 97.5% two-sided CI for the serotype-specific IgG GMC ratio (test group to control group) is above 0.5 or not. To take account of α inflation and β consumption, Bonferroni correction will be employed to α, producing corrected α=0.025/2=0.0125 and corrected power=1–0.2/7=0.97.
For each of the six additional serotypes in PCV13 only: On the basis of the preclinical research, we hypothesise that the proportion of vaccine recipients reaching the serotype-specific IgG concentration threshold of 0.35 μg/mL in the test group will be about 80% while that in the control group will not be >50%. If there is a notable difference between the two groups, a sample size of 274 infants per group, 548 total, will provide 80% power to demonstrate that the lower limit of 95% two-sided CI for a proportion of the test group will be no <70% (the target rate). To control for family-wise type II error, power will be adjusted to 0.97 (equals to 1–0.2/6).
Final decision: Theoretically, a total of 868 evaluable participants (434 per group) should enable the trial to achieve the primary outcomes. To take blood draw failures and blood collection refusals into consideration, we will allow for an anticipated 20% dropout rate.20 Therefore, the total sample size required is 1040 infants (520 per group).
Randomisation and allocation
The allocation sequence is generated by the Department of Health Statistics of Fourth Military Medical University using the SAS (SAS Institute , Cary, North Carolina, USA) Plan procedure. In order to guarantee the group balance in different sites, the block randomisation kit will be used. Each site will receive consecutively coded vaccines.
Owing to the difference in appearance of PCV13 and PCV7, the administrators cannot be blinded, but the group assigners, laboratory staff and investigators will be. Before recruitment, all vaccines will be masked in identical white small boxes and labelled with vaccine numbers based on the allocation schedule by those who are not involved in the clinical trial procedures. Each vaccine number is composed of two parts: the subject identification number and the dose specification number. Four vaccines with the same subject number prefix will belong to the same participant. In case any unexpected situations arise, additional vaccines including PCV13 and PCV7 in a ratio of 2:1 will be prepared and wrapped separately in individual white boxes labelled with numbers from B0001 to B0540. The link between the additional vaccine number and subject number will be established based on an allocation sequence list by Medidata Rave—the electronic data capture (EDC) system for this study. Emergency letters will be sent to each of the sites and properly preserved until needed.
Each eligible participant will be sequentially allotted a unique subject number according to the site allocation list by authorised assigners, and thus will have an equal chance of being assigned to either the test group or the control group. Blood samples will be labelled with subject numbers and collected independently of the vaccination process. The inoculator is responsible for taking out the vaccine related to a specific subject number and administering the injection. Inoculators must not share any allocation information with investigators or those involved in follow-up procedures.
Withdrawal
The participants’ parents or legally responsible representatives will be aware that their decision to take part in the study is voluntary and that they are free to withdraw consent at any time. All withdrawals from the randomised intervention will be reported. The investigator may drop participants during the study in the case of intercurrent illness, AEs, SAEs, protocol violations or other reasons that meet the exclusion criteria.
Data management
The trial data will be managed using the Medidata Rave EDC system. The database software provides several features to help maintain data quality. These features include, but are not limited to, keeping audit trails on all data fields, allowing custom functions in configurations and opening both automatic and manual queries. At the same time, the original record cards will be designed in accordance with the protocol to collect raw clinical data and to support the trial procedures. The source data obtained from each participant will be examined and verified by investigators and clinical research assistants (CRAs) at each study site. The entered data will be systematically validated and audited by the EDC system itself using edit checks, as well as by CRAs and data managers (DMs). Data query forms generated by medically trained personnel will be sent to the site investigators for identification. The electronic-case report form (e-CRF) will be inspected by DMs at regular intervals throughout the trial to verify the protocol compliance. The entered data should be complete, consistent and accurate. The blinded immunology assay results will be sent in an additional Excel spreadsheet via email to the data manager to be linked with the central database.
Statistical methods
Data set: The full analysis set (FAS) will comply with the principles of intent-to-treat (ITT)21 and comprise all participants who receive at least one dose of vaccine and one follow-up visit. The per-protocol analysis set (PPS) will comprise ITT participants who have received the integrated vaccination schedule and participated in all follow-up visits. Whether a participant with protocol deviations should be eliminated from the PPS set will be determined through blind data review process before analysis. The safety analysis set will comprise participants who were randomised to a certain group and vaccinated including those with protocol deviations.
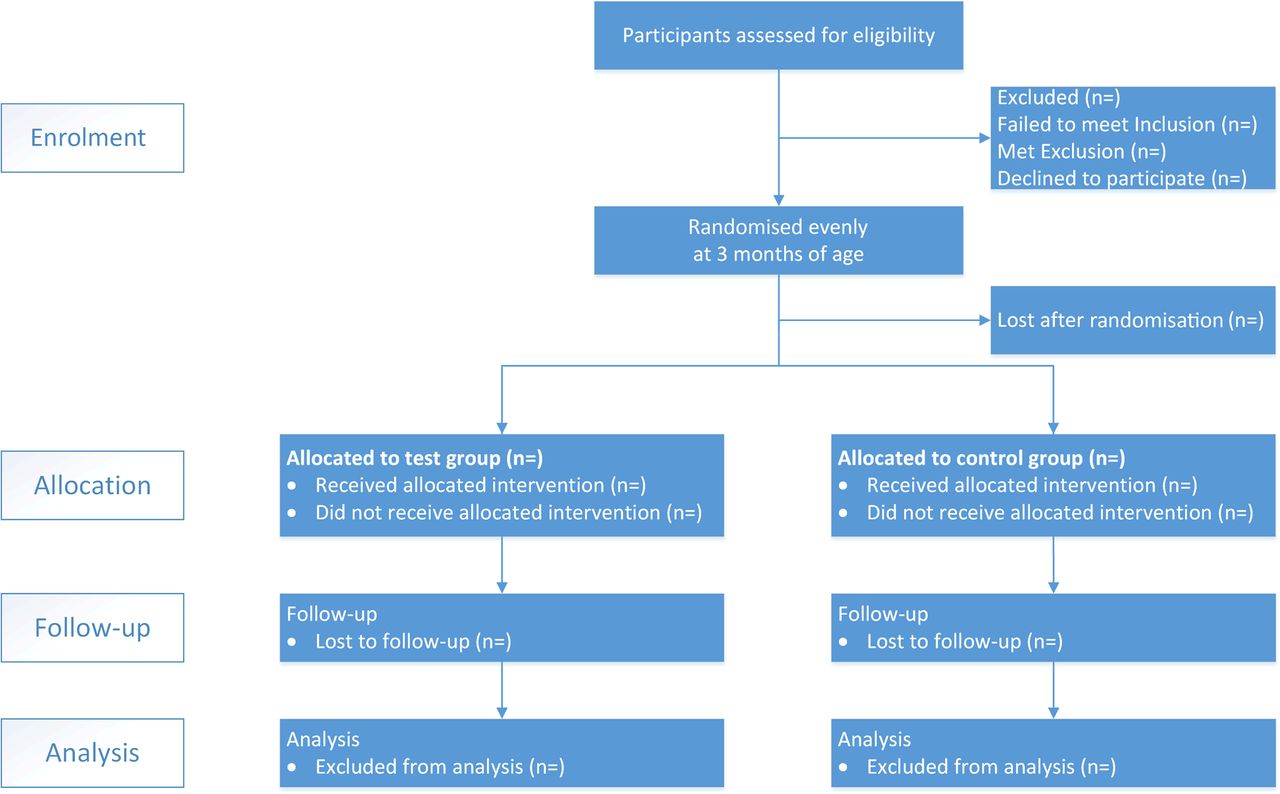
Analysis principle: Analysis will be conducted following the CONSORT22 flow diagram (figure 1) of the phases of the study: enrolment, allocation, interventions, follow-up and analysis. Baseline characteristics analysis will be given for each group to depict the premises of this study using means, SD, minimum and maximum value as well as the two-sided 95% CIs (if any). Continuous data will be portrayed with means, SDs and CIs if a normal distribution hypothesis of data is met. Binomial data will be summarised as frequencies and proportions. All statistical tests will be demonstrated with a two-sided p value, and all CIs will be calculated as 95% at a two-sided level unless otherwise specified. A detailed statistical analysis plan will be finalised before the first substantive statistical analysis.

The study flow diagram.
Immunogenicity: The immunogenicity population consists of all participants who belong to PPS and have available serological data before and after immunisation. The primary immunogenicity end points for each of the pneumococcal serotypes are the proportion of participants reaching the serotype-specific IgG concentration threshold of 0.35 µg/mL, and the geometric mean IgG antibody concentration, in the PCV13 group measured 30 days after the primary series. Within each group, for each pneumococcal serotype, the proportion of participants with a specific-IgG antibody concentration threshold of 0.35 μg/mL and the two-sided 95% CI for the proportion will be calculated; for each antibody concentration, the value will be logarithmically transformed to base 10, and the two-sided 95% CI will be structured. To evaluate differences between two groups, the sequential test (figure 2) of hypotheses, including the non-inferiority for the seven common serotypes and the efficacy for the six additional serotypes, will be performed. The non-inferiority for each of the seven common serotypes will be declared if the lower limit of the two-sided 97.5% (one-sided 98.75%) CI, which is calculated using the exact binomial method23 ,24 for the difference in proportions (PCV13—PCV7), is no <−10%, or the lower bound of the two-sided 97.5% (one-sided 98.75%) CI of the IgG GMC ratio (PCV13/ PCV7) is >0.5. To control for familywise type I error, Bonferroni correction will be used to address multiple test, and thus a two-sided significance level of 0.025 (0.0125, one-sided) will be adopted. Only after each of the seven common serotypes has been shown to be non-inferior will the other six additional serotypes be worth assessing. The efficacy criteria for each of the six additional serotypes will be met if both the lower limit of the two-sided 95% CI for the proportion of vaccine recipients reaching the serotype-specific IgG concentration threshold of 0.35 μg/mL conducted by the normal approximation method is no <70%, and the two-sided 95% CI for the difference in proportion (PCV13–PCV7) compared by a χ2 test lies almost to the right of the zero (zero or positive mean difference). A p value of <0.05 is considered statistically significant.

{kind=link}
{kind=link}
The sequential test flow chart. GMC, geometric mean concentrations; PCV13, 13-valent pneumococcal conjugate vaccine; PCV7, 7-valent pneumococcal conjugate vaccine.
The secondary immunogenicity end points for each of the pneumococcal serotypes in the PCV13 group are the percentage of vaccine recipients reaching a serotype-specific IgG concentration threshold of 0.35 µg/mL 30 days after the toddler dose and the percentages of vaccine recipients reaching a serotype-specific IgG concentration threshold of 1.0 µg/mL 30 days after the primary series and booster dose. The proportions of the participants reaching the OPA titre threshold of 1:8 after primary and booster immunisation in predefined subgroups also serve as the secondary immunogenicity end points. Within each group and for each antibody concentration or OPA titre, geometric mean will be calculated and each concentration or titre will be logarithmically transformed for analysis. To assess differences between the two groups and to test the non-inferiority of the PCV13 group, the analysis method described in the primary end points section will be employed.
As reported in previous studies,25 ,26 antibody responses to serotypes 19A, 6A and 5 were noted in the PCV7 group. Therefore, when evaluating the efficacy of 6A, 19A and 5 in the PCV13 group, the OPA results should be combined.27
Safety: The safety population includes all participants in the safety analysis set who will receive at least one dose of the study vaccine. Safety analysis including comparisons of incidences of local reactions, systemic events, use of concomitant medications, dropouts and AEs between PCV13 and PCV7 will be demonstrated with the corresponding two-sided 95% CIs and p values base on Fisher's exact test methodology for each dose, respectively, and for all in total as well. A p value equal to or <0.05 indicates a statistically significant difference.
Harms
Any SAEs will be reported to regulatory authorities, sponsors and Ethics Committees (ECs) within 24 hours by fax, email or in person. Any suspected or unexpected serious events will be reported to regulatory authorities by sponsors within seven calendar days and its follow-up investigation report will be submitted within eight calendar days of the first report submission day.
Auditing
Monitoring of this trial will be conducted by a Contract Research Organisation (CRO, Simoon Record Pharma Information Consulting Co). Guidelines of Good Clinical Practice (GCP) will ensure compliance.
Dissemination
Informed consent
All participants' parents/legally responsible representatives will receive adequate information about the nature, purpose, and possible risks and benefits of the trial by an informed consent form approved by the EC. The original version of signed consent forms will be retained by the investigators as a trial record, and a copy will be kept by the participants' parents/legally responsible representatives.
Confidentiality
To assure confidentiality, all participants will be allotted a unique subject number throughout the trial. Personal information will be collected with standardised paper forms by face-to-face interviews. Paper-based data will be stored in locked cabinets with limited access at each site, and the electronic data will be saved on password protected computers.
Access to data
The final data set will be under the custody of the sponsor together with the CRO selected beforehand. The data managers will be permitted to access the full analysed final data set. Access to the final or identifiable data by others is subject to the sponsor's approval.
Dissemination policy
The trial will be reported in accordance with the CONSORT guidance.
Discussion
Until now, the efficacy and safety of PCV13 among the Chinese population are still largely unknown. Hence, this study intends to characterise the PCV13 among Chinese infants and develop a safety profile for the vaccine. To fulfil these objectives, we calculated sample sizes under different hypothetical conditions and determined the optimal sample size to use to take all factors into consideration. In study design, we adopted the sequential test method to inspect the non-inferiority of each of the seven common pneumococcal serotypes and the efficacy of each of the six additional pneumococcal serotypes step by step to avoid false-positive error inflation. To be more discreet and conservative, instead of setting pneumococcal serotype 6B in PCV7 as a baseline when verifying the immunogenicity of each of the six additional serotypes in PCV13, as many other researchers have done, we defined 70% as the lower limit of the two-sided 95% CI for the percentage of responders that reach the serotype-specific IgG concentration threshold of 0.35 μg/mL and supposed that the corresponding percentages in PCV7 are <50%.29 Allowing for the cross-reactivation between 6A and 6B as well as 19A and 19B in PCV7, we will select a subgroup of which to obtain the OPA results for each of the 13 serotypes to correct for potential bias.
Given the main focus of the research, this study is not designed to involve the assessment of concomitant immunisation with other vaccines specified in paediatric vaccination schedules such as diphtheria, tetanus and acellular pertussis vaccine (DTaP), which might be regarded as a potential limitation. Since the feasibility of PCV7 administered concomitantly with DTaP in mainland Chinese infants has been shown in the research reported by Li RC et al,30 and the immunogenicity of PCV13 coadministered with DTaP was demonstrated by Togashi T et al,7 we will not prohibit the usage of vaccines listed in the expanded programme on immunisation, but we will compulsively demand at least a fortnight's interval between two kinds of vaccination.
References
Footnotes
Contributors JLX provided statistical expertise and created the study design together with JJC. ZH and LY conceived the study and participated in study design. JJC drafted the manuscript and calculated the sample size. NMS, YLZ, SLX and GHL are the primary investigators and helped with revising the protocol. RCL and YPL are the senior consultants and contributed to protocol development. SYY compiled the original protocol in Chinese and participated in study design discussion. All authors read and approved the final manuscript.
Funding The study is supported by a grant from Bio-pharmaceutical Major Scientific and Technological Project (biological product development section) of Yunnan Province (project number 2015ZJ005). The funding source has no role in the trial design and will have no role in the trial conduct, data analysis and interpretation, or writing or reporting.
Disclaimer The views expressed are those of the author(s) and not necessarily those of the DHSFMMU, Walvax or the Centre for Disease Control and Prevention.
Competing interests We have read and understood BMJ policy on declaration of interests and declare the following interests: This trial is sponsored by Walvax Biotechnology Co. Walvax, as the sponsor, is responsible for organising and managing the study to support the prelicensing evaluation of the investigational PCV13. The new PCV13 was researched and developed by Walvax and manufactured by an affiliate of Walvax. JJC, LY, ZH and SYY work at Walvax and belong to the clinical research team of PCV13.
Ethics approval The trial is approved by the Beijing Chaoyang District Centre for Disease Control and Prevention Ethics Committee (the lead institution), the Heibei Province Centre for Disease Control and Prevention Ethics Committee, the Henan Province Centre for Disease Control and Prevention Ethics Committee, and the Shanxi Province Centre for Disease Control and Prevention Ethics Committee. The registration number of this trial is NCT02736240.
Provenance and peer review Not commissioned; externally peer reviewed.