Article Text

Abstract
Introduction Some evidence indicates that exogenous surfactant therapy may be effective in infants with acute viral bronchiolitis, even though more confirmatory data are needed. To date, no large multicentre trials have evaluated the effectiveness and safety of exogenous surfactant in severe cases of bronchiolitis requiring invasive mechanical ventilation (IMV).
Methods and analysis This is a multicentre randomised, placebo-controlled, double-blind study, performed in 19 Italian paediatric intensive care units (PICUs). Eligible participants are infants under the age of 12 months hospitalised in a PICU, suffering from severe acute hypoxaemic bronchiolitis, requiring IMV. We adopted a more restrictive definition of bronchiolitis, including only infants below 12 months of age, to maintain the population as much homogeneous as possible. The primary outcome is to evaluate whether exogenous surfactant therapy (Curosurf, Chiesi Pharmaceuticals, Italy) is effective compared with placebo (air) in reducing the duration of IMV in the first 14 days of hospitalisation, in infants suffering from acute hypoxaemic viral bronchiolitis. Secondary outcomes are duration of non-invasive mechanical ventilation in the post-extubation phase, number of cases requiring new intubation after previous extubation within 14 days from randomisation, PICU and hospital length of stay (LOS), duration of oxygen dependency, effects on oxygenation and ventilatory parameters during invasive mechanical respiratory support, need for repeating treatment within 24 hours of first treatment, use of other interventions (eg, high-frequency oscillatory ventilation, nitric oxide, extracorporeal membrane oxygenation), mortality within the first 14 days of PICU stay and before hospital discharge, side effects and serious adverse events.
Ethics and dissemination The trial design and protocol have received approval by the Italian National Agency for Drugs (AIFA) and by the Regional Ethical Committee of Verona University Hospital (1494CESC). Findings will be disseminated through publication in peer-reviewed journals, conference/meeting presentations and media.
Trial registration number Clinicaltrials.gov, issue date 22 May 2019. NCT03959384.
- paediatric intensive & critical care
- paediatric infectious disease & immunisation
- paediatric thoracic medicine
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- paediatric intensive & critical care
- paediatric infectious disease & immunisation
- paediatric thoracic medicine
- clinical trials
Strengths and limitations of this study
This is the first large multicentre randomised trial to investigate the effectiveness and safety of exogenous surfactant in infants with severe viral bronchiolitis.
The main strength is the randomised and double-blind trial design, which will provide evidence about the therapeutic role of exogenous surfactant versus placebo (air) in severe cases of bronchiolitis requiring invasive mechanical ventilation.
Several Italian paediatric intensive care units will participate to this trial, enabling greater generalisability.
Another strength is the age limit we chose as entry criterion, by considering only bronchiolitis in patients aged less than 12 months, thus reducing the risk of confounding diagnoses, such as asthma-related obstructive diseases.
One limitation is that early diagnosis of bronchiolitis at enrolment is made by clinical examination, which may limit the diagnostic accuracy.
Introduction
Viral bronchiolitis is a common cause of hospitalisation for acute respiratory insufficiency in infants, especially in the first months of life.1 Despite several randomised controlled trials (RCTs) have tested the effectiveness of various agents, such as corticosteroids, inhaled beta-agonists, epinephrine, hypertonic saline, among others, no proven specific therapy for bronchiolitis is currently available.2–4 Recommended treatment remains essentially supportive, ensuring hydration and oxygen supplementation as needed.5–7 However, intensive care may be required for most severe cases, including invasive and non-invasive mechanical support, high-frequency oscillatory ventilation (HFOV), heliox, neurally adjusted ventilatory assist (NAVA) or extracorporeal membrane oxygenation (ECMO).8–12 Recently, Boet et al13 hypothesised a therapeutic role of exogenous surfactant in severe bronchiolitis. Indeed, endogenous surfactant plays a fundamental role in maintaining adequate surface tension in the small airways, thus preventing their collapse. Consequently, an alteration of surfactant components may increase submucosal oedema, mucus production and desquamation of airway ciliated cells, contributing to small airway obstruction.14 15 All this implies a functional impairment of bronchioles, which may cause excessive work of breathing, hypoxia and hypercapnia, sometimes requiring invasive mechanical ventilation (IMV).16
In 12 infants with viral bronchiolitis requiring intubation, Dargaville et al14 observed an impaired functional activity of endogenous surfactant, which resolved after clinical healing of the disease. Subsequent studies reported alterations in both surfactant proteins and pulmonary surface tension, using the ‘pulsating bubble surfactometer’ method or the ‘click test’ in tracheal aspirates obtained from children with bronchiolitis.17 18 Recently, Barreira et al19 reported qualitative and quantitative alterations in the lipid profile of surfactant in bronchiolitis caused by respiratory syncytial virus (RSV). Similarly, in a cross-sectional controlled study on 32 mechanically ventilated infants, including 16 with bronchiolitis and 16 controls with normal pulmonary function, Hartmann et al20 observed marked alterations of surfactant in patients with bronchiolitis, by using the count of lamellar bodies and the microbubble test.
Early studies have shown some positive effects of surfactant replacement in severe bronchiolitis. In a small study, Tibby et al15 assessed the effects of exogenous surfactant in 19 young infants requiring mechanical ventilation for severe RSV bronchiolitis. Nine infants received two doses of bovine surfactant (Survanta, 100 mg/kg) within 24 and 48 hours of mechanical ventilation, while 10 infants received placebo (air). The ventilation index (VI) and the oxygenation index (OI) significantly improved at 60 hours in the surfactant group. Furthermore, pulmonary compliance and airway resistance progressively deteriorated in the placebo group, but not in the surfactant group. However, the average ventilation time and paediatric intensive care unit (PICU) length of stay (LOS) did not differ between the intervention and control groups.15 In a small multicentre RCT, Luchetti et al21 assessed the efficacy of surfactant (Curosurf 50 mg/kg) in 20 ventilated infants with RSV disease, reporting an improvement in gas exchange and respiratory mechanics, as well as a shorter duration of mechanical ventilation and PICU LOS when compared with 20 controls. A recent meta-analysis evaluated the effect of natural surfactant in 79 infants with bronchiolitis requiring mechanical ventilation. Two trials did not use a placebo in the control arms, while the third used air as placebo.22 Interestingly, outcomes including the PICU LOS, PO2/FiO2 and CO2 elimination were better in patients treated with surfactant, with a marked trend toward less duration of mechanical ventilation. However, due to methodological flaws and the small sample, data were insufficient to confirm or refute the efficacy of surfactant therapy.22 Of note, no adverse effects or complications were reported in the surfactant-treated group.
Thus, there is a need for further randomised studies, adequately powered and in larger populations, to assess the efficacy and safety of exogenous surfactant therapy in infants with severe bronchiolitis.
To this end, the present study will evaluate whether Curosurf treatment is effective, compared with placebo (air), in reducing the duration of IMV in the first 14 days of hospitalisation, in infants under the age of 12 months with severe hypoxaemic viral bronchiolitis. Secondary outcomes are listed in table 1. Among them, we will assess whether Curosurf treatment is effective compared with placebo in reducing the duration of non-invasive mechanical ventilation in the post-extubation phase, the number of cases requiring reintubation, the PICU and hospital LOS, the duration of O2 dependency. Furthermore, we will assess the safety and tolerability of Curosurf in these patients.
Secondary efficacy, safety and tolerability endpoints
Methods and analysis
Study design
This is a multicentre randomised, placebo-controlled, double-blind study. Participants in the intervention group will be given exogenous surfactant (Curosurf), while those in the control group will receive placebo (air). The trial design is summarised in figure 1.
Setting
This study will be conducted in Italy, and participants will be recruited in 19 PICUs, in academic and community urban sites throughout the northern, central and southern regions of the Country.
Study population and eligibility criteria
The study population will comprise infants hospitalised in PICU for severe acute hypoxaemic bronchiolitis, requiring IMV. To maintain the population as homogeneous as possible, we adopted a more restrictive international definition of bronchiolitis, including only infants below 12 months of age.
Eligibility criteria
Each centre will screen subjects for eligibility criteria. Centres will compete to enrol the prespecified number of trial participants. Patients may be recruited either by the physician on duty or by a medical investigator. Differently, enrolment will be finalised only by a medical investigator, after checking eligibility criteria and after obtaining the signature of informed consent from both parents. Study inclusion and exclusion criteria are outlined in table 2. Withdrawal of parental consent will be a criterion for exiting the study.
Study participant inclusion and exclusion criteria
Randomisation
Participants will be randomly assigned either to intervention (group 1—Curosurf) or control (group 2—placebo) group, with a 1:1 allocation as per a computerised method of block randomisation between the two groups (software STATA V.11). To reduce predictability of a random sequence, details of block randomisation will not be available to those who enrol participants or assign interventions. The randomisation list will be built by the study coordinator centre. During the enrolment of any patient, a medical investigator of each centre will be allowed to insert the required patient’s data in an electronic clinical report form (e-CRF), immediately receiving both ID and treatment assigned to the patient.
Blinding
The study is double-blind, as the patient and patient’s parents on one side, and doctors and nurses who will be responsible for the patient’s care on the other side, will not be aware of the assignment of treatment. Then, at each centre only one physician and one nurse will know, prepare and administer the intervention (drug or placebo) for any enrolled patient, being subsequently not clinically involved in the care of that particular patient until discharge. The assignment of the type of treatment will be confidentially communicated by the coordinator centre to a medical investigator attending the patient’s bed. Then, the same medical investigator will take care of administering the treatment assigned to the patient, masking the procedure with appropriate precautions, for example, by using screens or closing the patient’s room, as appropriate. The preparation and administration of treatment, medication or placebo, will be done by a nurse who will not have to disclose the type of treatment assigned. Thereafter, such a nurse will not be involved in the patient’s care until the conclusion of the study. Instead, the patient evaluation will be carried out by other physicians and/or nurses who will not be aware of the type of treatment carried out. Despite that the execution and maintenance of blinding can cause some difficulties, we aim to reduce the bias effect that medical and nursing staff might have, being aware of the therapeutic arm applied to that single patient.
Unblinding will not be permissible unless exceptional circumstances, when knowledge of the actual treatment would be deemed as absolutely essential for further management of the patient. Given the design of this study and the safety profile of the drug, we do not foresee specific harms for the patient, severe enough to justify a code break. Furthermore, unblinding should not necessarily be a reason for study drug discontinuation (eg, cancelling the second intervention dose, if indicated by the study protocol).
Intervention
Early after randomisation, the assigned treatment will be carried out in two phases, to be performed in rapid sequence:
Group 1: Curosurf
The patient dosage is 50 mg/kg of Curosurf, to be split into two units of 25 mg/kg each. The dosage of 50 mg/kg is supported by previous studies that had employed the same type of surfactant (Curosurf).21 23
For phase (1), a bronchoalveolar lavage (BAL) will be performed with 25 mg/kg of Curosurf, diluted 1:10 with normal saline, divided into two aliquots administered through the endotracheal tube in two different postures (first BAL on right decubitus, second BAL on left decubitus). A sterile gastric tube of non-obstructive size will be connected to a syringe containing the diluted solution. The gastric tube will be inserted within the tracheal tube to facilitate the drug instillation. If needed, pre-oxygenation will be given just before the procedure, at 0.3–0.6 fraction of inspired oxygen (FiO2), titrated to maintain patient’s oxygen saturation >95% for at least 30 s. For phase (2), immediately after phase 1, surfactant supplementation will be provided with a further dose of 25 mg/kg of Curosurf, diluted with normal saline 1:2 (1 mL=40 mg of surfactant) and administered in two aliquots through the endotracheal tube in two different postures (first on right decubitus, second on left decubitus).
Group 2: placebo (air)
Ambient air aspirated in sterile syringes will be used as placebo, adopting the same procedure followed for the administration of the intervention drug. The choice of air as placebo is supported by previous studies testing surfactant treatment in paediatric patients with bronchiolitis or acute lung injury.15 24 The use of substances other than ambient air, such as normal saline, would not be ethically correct, given the possibility of adverse effects on the patient (eg, removal or altered functionality of native surfactant, airway obstruction or vagal stimulus).
While administering ambient air through the tracheal tube via a sterile gastric tube, sequence and timing adopted in group 2 will be identical to those used in group 1. In particular, patients will be moved from right decubitus to left decubitus for the first and the second administration. The aliquots of air will correspond to the volume of the diluted solutions used for group 1.
Repetition of treatment (Curosurf or placebo)
The same assigned treatment (either Curosurf or placebo) will be repeated at least 12 hours after, and within 24 hours from the first treatment, using the same dosage and the same procedure for administration, if at least one of the following indications is present:
OI >8 or an oxygen saturation index (OSI) >7.5.
FiO2>40% and PEEP >5 to maintain oxygen saturation (SpO2) >95%.
The replication of treatment will follow the same modalities of the initial treatment in both groups. The medical researcher repeating the second treatment will be the same who administered the first one. Should this not be possible, for organisational or shift reasons, the medical researcher will communicate the type of treatment to be repeated to a second researcher, confidentially. If needed, this second physician will, in turn, involve another nurse to perform the procedure, binding the nurse not to disclose the treatment administered to the rest of the team. Thereafter, such a nurse will not be involved in the patient’s care until the conclusion of the study.
Evaluation during the treatment period and patient parameters collection
Regardless of the type of treatment received, all enrolled patients will be assisted according to the normal clinical practice of each centre. In addition, several parameters will be collected 15 min before, and 2, 6, 12, 24, 36 and 48 hours after administration of the drug: OSI and OI, tidal volume (TV), positive end-expiratory pressure (PEEP), peak inspiratory pressure (PIP), respiratory frequency, FiO2, inhalation time (Ti), mean airway pressure (MAP), PaCO2, end tidal CO2. Should the treatment be repeated, all parameters will be recollected according to the same time points. Study data will be collected by research assistants and entered into the e-CRF, a secure, web-based application hosted by CRT (Clinical Research Technology), ad hoc designed for the present study. Data will be also registered in the clinical chart of each patient. Assessors will receive training for ensuring the quality of trial data. In each centre, data entry and management will be under the local principal investigator (PI) responsibility. Original data will be kept within the clinical chart of each patient. A full copy of the chart will be safely stored in a secure and accessible place by the local PI. Participant files will be kept in storage for 5 years after completion of the study.
Control arm and interventional arm
All enrolled subjects will be treated according to the standards of good clinical practice, with special attention to sedation and analgesia of patients in PICU, as well as taking care of and supporting their family members. During the trial, relevant concomitant care and interventions, such as HFOV, iNO or ECMO, will be permitted, according to local protocols. Their use within 14 days of randomisation should be reported in the eCRF with the specific date.
Finally, the possibility of suspending oxygen therapy, extubating the patient, discharging from the PICU and the hospital will be assessed at least daily, according to local practices. Dates and time of these events will be reported in the eCRF, if they occur within 14 days of randomisation. The date of hospital discharge or exitus will be also collected.
Safety monitoring
Any adverse event (AE) or serious adverse event (SAE) will be reported on the original documentation and in the specific sections of the eCRF. Any AE, which at the time of the first registration are not resolved or permanently stabilised, shall be followed until the end of the study, providing the appropriate updates. An AE/SAE with a highly probable/probable/possible causality with the intervention drug is considered to be related to the latter and therefore will assume the definition of adverse drug reaction (ADR). Any SAE and serious adverse reaction will be verified by the Data Monitoring Commission and reported to the competent authorities. The Pharmacology Service of the University Hospital of Verona is responsible for the notification of SUSAR in Eudravigilance following the applicable regulations for non-profit intervention studies. In case of AE and/or serious and non-serious adverse reactions, each investigator must send a copy of the CRF data collection-adverse events to the pharmacology service of the University Hospital of Verona. Once a year and for the duration of the clinical trial, the Pharmacology Service will send the Development Safety Update Report (DSUR) to the Ethics Committee and to the AIFA (Det. AIFA No. 9/2012).
Interim safety analysis
To assess safety, a single interim analysis has been programmed, at a time point where about half of the subjects will have completed the study (80 patients). Therefore, it will not be necessary to revise the sample number according to such analysis.
Data management and procedures to ensure the confidentiality of data
To guarantee the confidentiality of data, each patient will be registered and deidentified by an identification code.
All the information concerning the enrolled subjects collected during the study, first on paper on the data collection card (CRF), will be transferred and recorded in a specific database with passwords, will be kept confidential and be treated in full respect of national Legislative Decree 196/03, in application of which all participants' parents will be asked to sign the informed consent. Access to data will be exclusively allowed to the PICU research team of the Department of Paediatric Critical care, University Hospital of Verona, as well as to the PICU Investigators from the other participating centres.
Study outcome assessment
As primary endpoint we will measure the number of IMV-free days from randomisation to day 14, considering the first successful extubation (at least 48 hours without the need for reintubation). We believe this endpoint reflects the importance of reducing the burden of prolonged IMV in these patients.
Secondary outcome measures will include the following:
Number of non-invasive mechanical ventilation-free days, from randomisation to day 14.
The necessity of reintubation, after previous extubation, within 14 days from randomisation.
(3.1) Number of PICU-free days, from randomisation to day 14; (3.2) total number of days of PICU stay from randomisation.
(4.1) Number of hospital-free days, from randomisation to day 14; (4.2) total number of hospitalisation days from randomisation.
Number of oxygen supplementation-free days, from randomisation to day 14.
OSI (or OI, if an arterial line is available) and VI values (PaCO2, end tidal CO2) detected 15 min before administration of treatment and at a distance of 2, 6, 12, 24, 36 and 48 hours.
Values of the mechanical ventilation parameters, such as TV, PEEP, PIP, respiratory frequency, FiO2, inspiration time (Ti) and MAP, detected 15 min before administration of treatment and at a distance of 2, 6, 12, 24, 36 and 48 hours after treatment.
Number of patients undergoing a repeated treatment (Curosurf or placebo) within 24 hours of the first treatment.
Number of patients undergoing non-conventional therapies (HFOV, ECMO, nitric oxide) during the first 14 days.
(10.1) Mortality during the first 14 days of hospitalisation; (10.2) mortality before hospital discharge.
Safety and tolerability outcome measures will include several secondary safety and tolerability endpoints, associated with the administration of assigned treatment within the first 48 hours after treatment:
Number of serious desaturation episodes (SatO2<75%) during the administration of treatment.
Episodes of severe bradycardia (FC<80/min) during the administration of treatment.
Number of episodes of extreme bradycardia or cardiac arrest with the need for chest compressions and/or administration of drugs for resuscitation (eg, epinephrine) during the administration of treatment.
Number of episodes of pulmonary haemorrhage during and within 48 hours after administration of treatment. Diagnosis of pulmonary haemorrhage will be confirmed by bloody endotracheal aspirate, radiographic findings or both.
Number of episodes of pneumothorax in the first 48 hours following treatment.
Sample size
Sample size calculation for the primary endpoint is based on historical data and expectations of the effect of surfactant therapy. Analysing a large national database (Italian TIPNET network) and selecting only those subjects with bronchiolitis who had a duration of IMV less than 15 days (about 240 px), we obtained a mean (±SD) duration of the IMV: 4.51±3.42 days (data not shown). Considering a level of significance of 0.05, a power of 0.80 and a common SD of 3425 days, 164 subjects (82 subjects per group) are required to identify a clinically significant difference of 1.5 days in the mean duration of IMV of group 1 compared with group 2, using a bilateral test. Assuming a dropout rate of 5%, the estimated number of subjects was 86 per group, with a total number of 172 enrolled participants. Calculations were performed with STATA (V.11). Despite a previous Cochrane meta-analysis resulted in a decrease in mechanical ventilation of about 63 hours,22 we conservatively hypothesised a smaller reduction in the duration of IMV (36 hours), as we thought this was more realistic and still meaningful from a clinical point of view.
Data analysis
The intention-to-treat (ITT) analysis includes all randomised subjects. The ITT population will be the primary population for the analysis and evaluation of primary efficacy. The population of per protocol (PP) analysis will include all ITT subjects without significant protocol violations. The analysis of effectiveness will be based on the population PP. All summary statistics will be provided according to the therapy group, including frequency and percentage (for categorical variables) and mean, median, SD, minimum and maximum (for continuous variables). The appropriate confidence intervals at 95% (95% CI) will also be calculated. For the analysis of the primary endpoint, the Student’s t-test will be used to verify any difference in the average number of days elapsed in IMV between group 1 and group 2. If the norms of normality and homoscedasticity are violated, the test of the sum of the ranks of Wilcoxon-Mann-Whitney will be used. A p value of less than 0.05 will be considered significant. The relative risk and the corresponding 95% CI will be calculated as indicated. Other endpoints will be analysed through the analysis of variance to repeated measures if all the assumptions will be verified; otherwise, the Friedman test or the mixed effects model will be used. For safety and tolerability endpoints, incidence and frequency of severe desaturation, bradycardia, pneumothorax and extreme bradycardia or cardiac arrest will be analysed.
For exploratory purposes, linear or logistic regression models will be estimated, depending on the nature of the independent variable, to verify the possible influence of variables such as age or basal FiO2/MAP values.
Data and safety monitoring committee
This committee will be composed by two physicians not involved, either directly or indirectly, in the treatment of any patient recruited. They will evaluate the results of the interim safety analysis and the evaluation of any SAE and/or serious adverse reaction that should occur throughout the duration of the study. The conduct of the trial is covered by an ad hoc insurance policy for the duration of the study in compliance with the DM 14/07/2009.
Ethics and dissemination
The trial design and protocol have received approval by the Italian National Agency for Drugs (AIFA) and by the Regional Ethical Committee of Verona University Hospital (1494CESC). Trial registration number NCT03959384; Pre-results. Table 3 summarises the study protocol and trial registration information.
Trial registration data: SURFABRON
Once a patient meets the inclusion criteria, the possibility to participate in this study will be proposed to his/her parents. Informed consent (approved by the local ethics committee) will be provided to parents and explained comprehensively by the principal investigator of each participating centre. The investigator will always be willing to answer any further questions that parents would want to ask before making a decision. The latest revision of the Declaration of Helsinki and the Oviedo Declaration are the basis for the ethical conduct of the study. The study protocol is designed and will be conducted to ensure adherence to the principles and procedures of Good Clinical Practice and to conform to the Italian laws. All data collected will be anonymised by the use of a numerical code without reference to the names of patients. Access to such data is only allowed to authorised personnel and directly involved in the study.
Dissemination
Study findings are expected in 2022 and will be presented at international conferences and submitted for publication in an international peer-reviewed journal. The criteria established by the International Committee of Medical Journal Editors will be adopted to publish the results of the research project in national or international scientific journals. The right to authorship will be based on the substantial contribution of the participants in the study to the analysis and interpretation of the data, drafting of the article and its critical revision and final approval of the document to be submitted for publication. Deidentified individual participant data for all primary and secondary outcome measures will be made available within 9 months of study completion, up to 5 years. Data access request will be reviewed by the steering committee. Requestors will be required to sign a Data Access Agreement. The Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) checklist for this protocol is located in the online supplemental appendix 1.
Supplemental material
Patient and public involvement
Viral bronchiolitis has been ranked as one of the most important causes of hospitalisation in young infants worldwide, causing a burden to their families. Patients’ caregivers were not involved in the design of the study. Ethical approval for this study was obtained from a board composed of professionals and lay men and women, also considering non-professional opinions. Results from this trial will be made available to public through communication also in non-specialised media.
Discussion
This will be the first large multicentre randomised trial to investigate the effectiveness and safety of exogenous surfactant in infants with severe viral bronchiolitis. There are potential advantages of surfactant therapy in these critically ill patients, including the following: (1) a shorter duration of endotracheal intubation and IMV, both factors being associated with short-term and long-term airways and lung injury; (2) a shorter LOS in highly specialised units, such as the PICUs, which are costly and burdened with higher risk of nosocomial infections; (3) an improvement of oxygenation and CO2 elimination in the acute phase of the disease. The main strength of this protocol is the randomised and double-blind trial design, which will provide evidence about the therapeutic role and safety of exogenous surfactant versus placebo, in patients with severe cases of bronchiolitis requiring IMV. The population age will be restricted up to 1 year, thus reducing the risk of confounding diagnoses, while the multicentre participation will enable a greater generalisability of the findings.
Trial status
Patient recruitment is in progress.

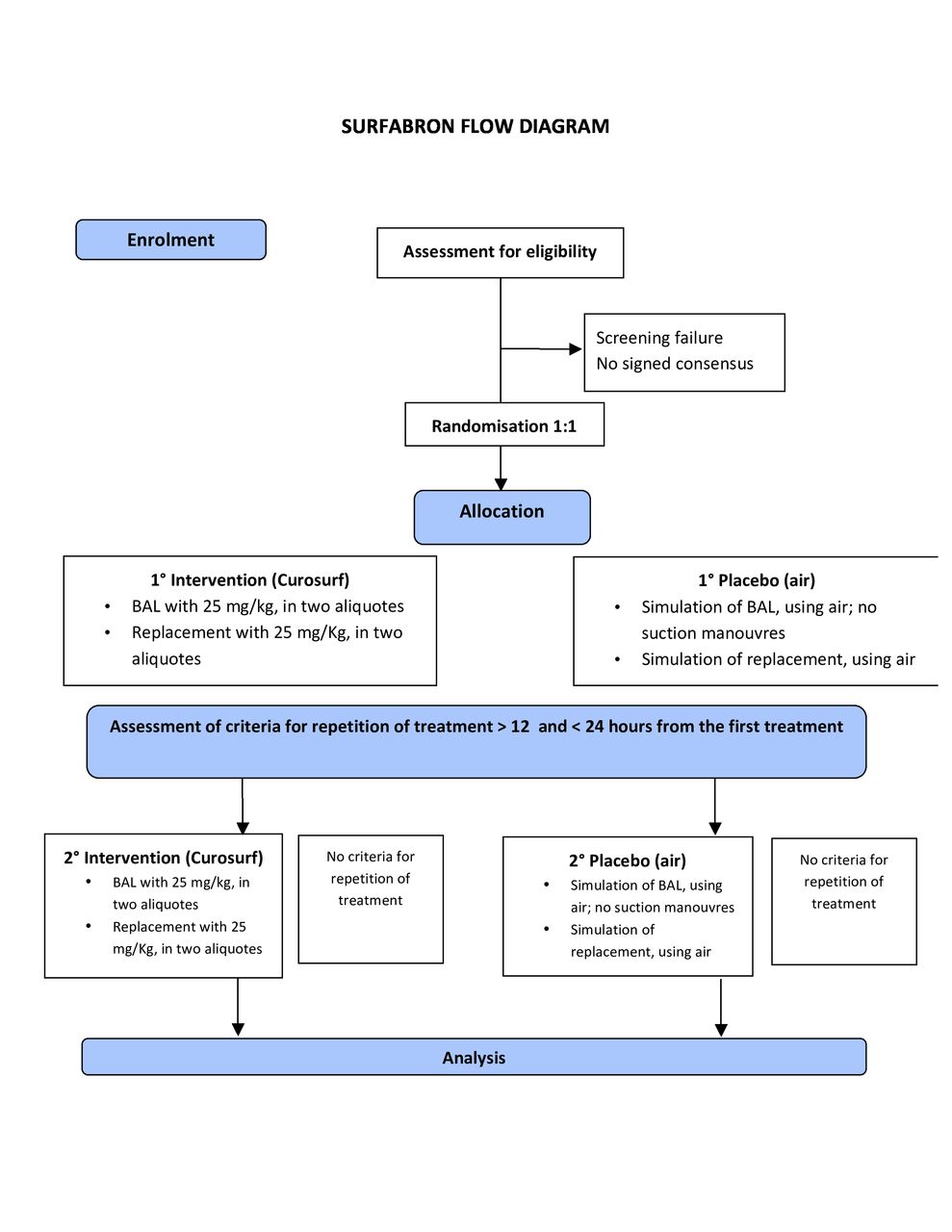{kind=link}
Flow diagram of the SURFABRON trial. BAL, bronchoalveolar lavage.
Acknowledgments
The authors would like to thank Ms CM for the statistical support, Mrs SM for refining the protocol structure, Ms FM for her support in contacting and managing all the participating centres.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors PB conceived the study and developed its protocol, wrote the first manuscript draft and subsequent revisions. GC and AMW revised the manuscript. SSC, EG, IR, AM, CM, FS, RS, GG, MB, FC, MF, AP, VS, LD, GZM, EC, SSC, AB, FT, EB, DB, GI, and PS are investigators and responsible for enrolment,determining treatment indications and data collection. All authors contributed to refinement of the study protocol and approved the final version. All the authors agreed to be accountable for all aspects of the work. There are no plans for the employment of professional medical writers.
Funding This study was sponsored by the Azienda Ospedaliera Universitaria Integrata Verona, that covered the cost for the electronic clinical report form (CRF). Chiesi Group covered the costs related to the insurance for the study. The sponsor and the funder were not involved in the study design and will not be involved in collection, management, analysis and interpretation of data, writing of the report and the decision to submit the report for publication.
Competing interests PB has received speaker fees from Chiesi Group. AW has received speaker fee from Takeda. FC and PS received travel funding by Chiesi. All other authors (SSC, EG, IR, AM, CM, FS, RS, GG, MB, MF, AP, VS, LD, GZM, EC, SSC, AB, FT, EB, DB, GI) report no competing interests.
Patient consent for publication Not required.
Ethics approval The trial design and protocol have received approval by the Italian National Agency for Drugs (AIFA) and by the Regional Ethical Committee of Verona University Hospital (1494CESC). Substantive protocol amendments will be reviewed by Regional Ethical Committee of Verona University Hospital and described in trial reports.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.