Article Text

Statistics from Altmetric.com
Over the last decade, we have witnessed a revolution in the understanding of primary cardiac arrhythmia syndromes. These remarkable advances stemmed from the discovery of mutations, primarily in ion channel genes, underlying a number of these disorders. Only 10 years ago the long QT syndrome (LQTS) was considered one disease entity, the Brugada syndrome had just been described, and the short QT syndrome (SQTS) had never been heard of. Moreover, it immediately became obvious that genetic heterogeneity in these disorders is not the exception but the rule.
The recognition of the genetic substrate underlying the inherited arrhythmia syndromes has provided remarkable insight into the molecular basis of cardiac electrophysiology, including the role of the various ion channels and mechanisms of arrhythmias. The availability of a genetic diagnostic test has added an important diagnostic tool, providing new opportunities for patient management such as early (presymptomatic) identification and treatment of individuals at risk of developing fatal arrhythmias. Studies into genotype–phenotype relationships, carried out mostly for the LQTS, have uncovered important gene specific aspects of disease and indicated that patient management must take the nature of the gene affected into consideration. Initial thoughts that the genetic dissection of these disorders would facilitate therapeutic management of these patients have, however, not been substantiated. On the contrary, the heterogeneous (genetic, pathophysiological) character of all primary arrhythmia syndromes precludes uniform treatment.
Herein we review these developments at large, with emphasis on the issues that are relevant for understanding the pathophysiology of these syndromes and daily management of affected individuals.
CARDIAC ION CHANNELS AND THE ECG
The cardiac action potential is mediated by the exceptionally well orchestrated activity of a diversity of ion channels (fig 1). Cardiac ion channels1 are protein complexes in the sarcolemma of cardiomyocytes which, via highly regulated opening and closing (gating), conduct a selective and rapid flow of ions through a central pore. Spatial heterogeneity of ion channel expression underlies the different action potential morphology of the different parts of the heart which in turn ensures a coordinated contraction.

Ionic currents contributing to the ventricular action potential (A) and schematic representation of a cardiomyocyte displaying (only) those proteins involved in the pathogenesis of inherited arrhythmia syndromes (B). In panel A, the action potential is aligned with its approximate time of action during the ECG. In panel B, ankyrin-B, an adapter protein involved in the long QT syndrome type 4, is not depicted.
The maintenance of normal cardiac rhythm is dependent on the proper movement of ions mediating the action potential in each cardiac compartment. The ECG is the result of the travelling electrical impulse as measured at the body surface. The width of the QRS complex is determined by the time interval between the depolarisation of the first and last ventricular cells, and the ST-T segment results from an inhomogeneous repolarisation in the specific layers (endocardial, mid myocardial, epicardial) of the ventricular wall. Abnormalities in ion channel function can have disastrous consequences that manifest themselves as ECG abnormalities and arrhythmias. These disorders of ion channels, commonly referred to as “cardiac channelopathies”, have been brought into focus in recent years as mutations in genes coding for specific ion channels were shown to underlie specific forms of heritable arrhythmogenic disorders occurring in the structurally normal heart. These include the LQTS, SQTS, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia (CPVT), and idiopathic atrial fibrillation. These disorders fall under the category of monogenic disorders—that is, disorders that follow a clear mendelian pattern of inheritance and are classified as autosomal dominant (usual), autosomal recessive (rare), or X linked (not described in inherited primary arrhythmia disorders).
LONG QT SYNDROMES
The long QT syndrome, estimated to affect 1 per 5000 individuals, is a repolarisation disorder identified by prolongation of the QT interval on the ECG. It has long been recognised as a familial disorder, frequently presenting in childhood with syncopal episodes and potentially lethal torsades de pointes tachyarrhythmias which occur in a significant proportion of untreated patients. An autosomal dominant form (Romano-Ward syndrome) and an autosomal recessive form (Jervell and Lange-Nielsen syndrome, also associated with deafness) have been clinically recognised.
Repolarisation is a delicate process depending on an intricate balance between inward currents (sodium, Na+, or calcium, Ca2+) and outward currents (potassium, K+) (fig 2). Shifts in the balance of these inward and outward currents can either increase or decrease the action potential duration with resultant QT interval prolongation or shortening, respectively. Inhomogeneous repolarisation ensues, predisposing the heart to potentially lethal ventricular arrhythmias.
{kind=link}
{kind=link}
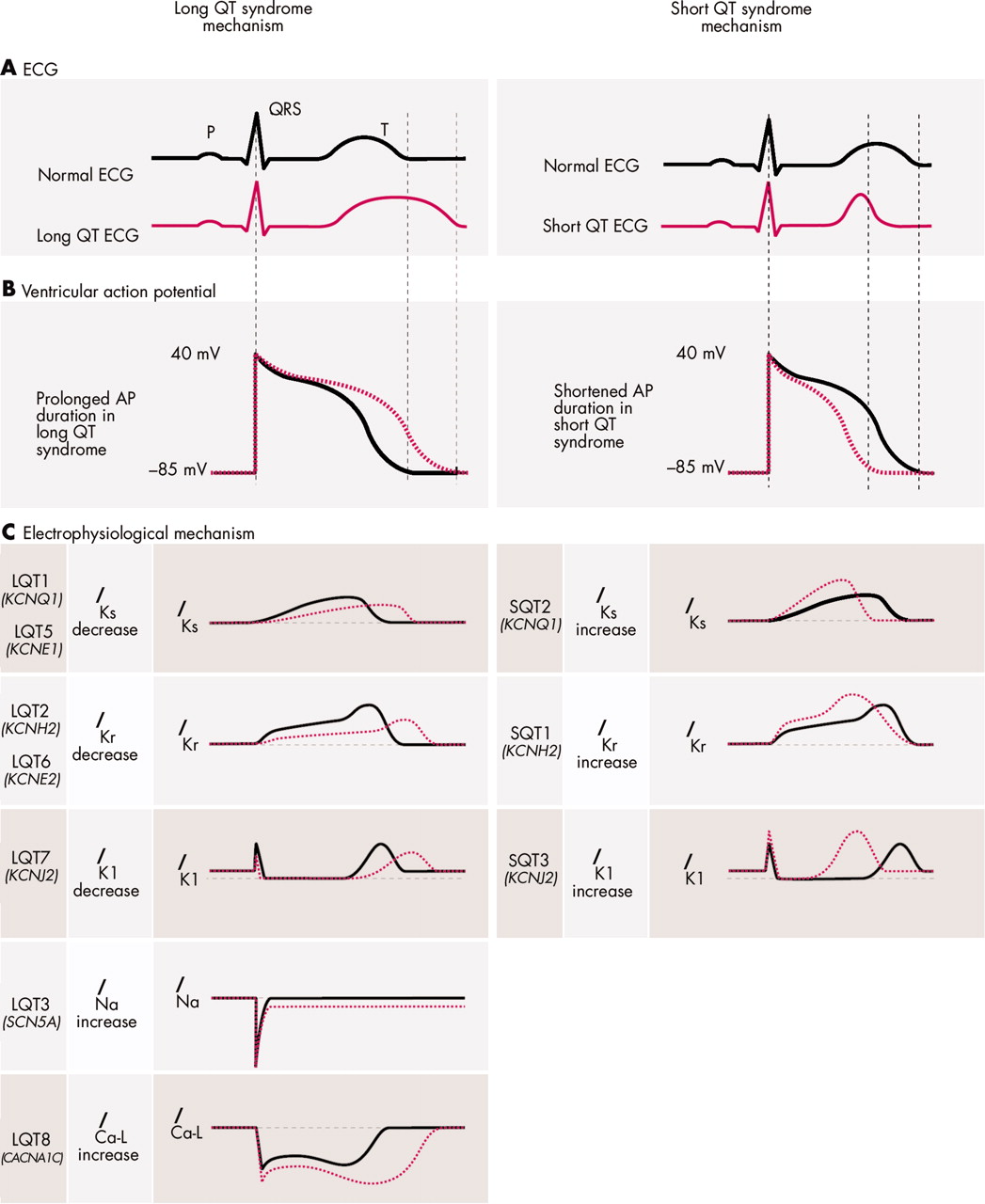
Pathophysiologic mechanisms of ion channel mutations in long QT syndrome (LQTS) and short QT syndrome (SQTS). (A) Schematic ECG representation. The red dotted line represents QT interval prolongation (left panel) and shortening (right panel) as can be observed in the LQTS and SQTS, respectively. (B) The ventricular action potential (AP). The red dotted line represents prolongation (left) and shortening (right) of action potential duration (APD) as can be observed in the LQTS and SQTS, respectively. (C) Effect of mutations on current characteristics in LQTS (left) and SQTS (right). By convention, upward (positive) deflections represent outward current, downward (negative) deflections represent inward current. Normal (solid grey line) and abnormal (dotted red line) current characteristics are aligned with their approximate time of action during the ECG (A) and the ventricular AP (B). A reduced IKs activity underlies APD prolongation in LQT1 and 5, while an increased IKs activity underlies APD shortening in SQT2. A reduced IKr activity underlies APD prolongation in LQT2 and 6, while an increased IKr activity underlies APD shortening in SQT1. A reduced IK1 activity during the terminal phase of repolarisation underlies APD prolongation in LQT7, while an increased IK1 activity underlies APD shortening in SQT3. An enhanced INa and an enhanced ICa-L during the AP plateau prolong APD in LQT3 and LQT8, respectively.
The common pathophysiological mechanism underlying QT interval prolongation in the LQTS is a net reduction in repolarising (that is, outward) current leading to an increase in action potential duration.2 Since K+ channels are the primary contributors to the repolarisation process it was no surprise that loss of function mutations in genes encoding for pore forming α-subunits (KCNQ1, KCNH2) of these channels and for their regulatory proteins (KCNE1, KCNE2) appeared to underlie LQTS (fig 2, left panel).2 Surprisingly, the SCN5A gene encoding the Na+ channel also appeared to be involved; gain of function defects in this channel lead to an increased inward Na+ current during the action potential plateau shifting the balance to prolonged repolarisation (fig 2).2
Another gene, ANK2, linked to LQTS, however does not code for an ion channel protein but encodes a membrane adapter protein, ankyrin-B.3 Mutations in ankyrin-B appear to lead to loss of function resulting in loss of expression and mis-localisation of the Na+/Ca2+ exchanger, the Na+/K+ ATPase, and the InsP3 receptor, with ensuing abnormal Ca2+ dynamics. This finding highlighted the fact that cardiac ion channels do not function in isolation, but form part of complex macromolecular assemblies (consisting of pore forming α-subunits, modulatory subunits, cytoskeletal and extracellular matrix elements, and associated signalling complexes) and that mutation in elements of this complex other than ion channel proteins per se could also lead to arrhythmias.
The autosomal dominant form of LQTS may be caused by mutation in any one of the above mentioned genes. However, mutations in KCNQ1, KCNH2, and SCN5A, underlying LQTS types 1, 2, and 3 (named in chronological order of identification, table 13–5w1–37), respectively, are by far the most common cause. The autosomal recessive form (Jervell and Lange-Nielsen syndrome) is caused by homozygosity (because of consanguineous parents) or compound heterozygosity for mutations in KCNQ1 or KCNE1. These genes are expressed in marginal cells of the stria vascularis where they are thought to play a role in the homeostasis of K+ in the endolymph, a K+ rich fluid of the inner ear. This explains the deafness in this disorder.
Summary of genes and chromosomal loci for inherited cardiac arrhythmia syndromes
LONG QT SYNDROME WITH EXTRACARDIAC FEATURES.
LQTS type 7, also known as Andersen syndrome, is a rare disorder with cardiac manifestations that include mild QT interval prolongation and significant U waves, frequent ventricular ectopy, bidirectional ventricular tachycardia (VT), and more rarely syncope, recurrent polymorphic VT, and cardiac arrest. The syndrome also exhibits extracardiac features including skeletal muscle periodic paralysis and developmental problems such as cleft palate, low set ears, short stature, and developmental features in the limbs.4 The different features of the phenotype are subject to significant variability in expression. Loss of function mutations in the KCNJ2 gene, which encodes the inward rectifier K+ channel Kir2.1 in both heart and striated muscle, has been linked to the disorder (fig 2).4 This channel conducts outward K+ current during the terminal repolarisation phase and diastolic phase of the action potential, thereby contributing to repolarisation and maintenance of membrane potential close to the resting membrane potential in both heart and striated muscle.
LQTS type 8 combines severe QT prolongation with syndactyly, baldness at birth, and small teeth in 100% of cases and less penetrant cardiac structural malformations, autism, mental retardation, and facial dysmorphic features.5 A single mutation in CACNA1C encoding the membrane Ca2+ channel accounts for all 13 cases described so far.5 This mutation leads to a gain of function defect, augmenting the depolarising current during the plateau phase of the action potential (fig 2), thereby prolonging the action potential duration. LQT8 is extremely rare and very malignant. Considering the widespread expression of the CACNA1C gene and the importance of Ca2+ as an intracellular signalling molecule, the widespread cellular and organ defects in this disorder are not unexpected.
SHORT QT SYNDROMES
The short QT syndrome is a recently described clinical entity that presents with a high rate of sudden death and exceptionally short QT intervals (QTc typically ⩽ 300 ms).6 So far only 30–40 patients have been described. Contrary to the LQTS, repolarisation is hastened by an enhanced outward current during repolarisation (fig 2). Gain of function mutations in the KCNH2 and the KCNQ1 gene, leading to an increase in outward K+ current through the respective K+ channels, were identified in patients with the disorder (table 1). Gain of function mutations in KCNJ2 also give rise to shortening of the QT interval, although not to the extent reported for KCNH2 and KCNQ1 mutations (table 1). In this genetic subtype, the terminal part of the ST segment is shortened giving rise to a peculiar ST segment.
BRUGADA SYNDROME
The Brugada syndrome was first described in 1992 and is characterised by ST segment elevation in the right precordial leads with or without conduction abnormalities, and a significant risk of sudden (nocturnal) cardiac death in the absence of structural heart disease.7,8 The electrocardiographic appearance may be variable and is under the influence of body temperature, autonomic transmitters, and drugs. The disorder is endemic in East and Southeast Asia, where it underlies the sudden unexpected death syndrome (SUNDS). Although the average age of patients experiencing arrhythmic events is 40 years, sudden death can strike at any age, even in very young children. Brugada syndrome is familial in about a third of patients, in which case an autosomal dominant mode of inheritance is observed. The SCN5A gene, which accounts for up to 20% of cases, is the only gene linked thus far to the disorder.8 The functional effects of Brugada syndrome causing SCN5A mutations are opposite to those found in LQTS. Thus, loss of function mutations underlie Brugada syndrome and the frequently associated (mild) conduction disorders.9 The pathophysiology of the typical ECG appearance is unresolved and may be caused by transmural differences in the action potential morphology (particular in the right ventricular free wall) or to local conduction delay, or both.10
CARDIAC CONDUCTION DEFECT
Cardiac conduction disease is mostly encountered as a consequence of cardiac injury (caused by ischaemia or surgery), as the major cardiac manifestation of neuromuscular diseases, or in association with congenital cardiac abnormalities. However, isolated cardiac conduction disease, with a progressive nature in some instances (also known as Lenegre and Lev disease) has also been described.
Three distinct genetic forms have been distinguished in families displaying an autosomal dominant inheritance (table 1). One form involves mutation in SCN5A. As for those causing Brugada syndrome, SCN5A mutations leading to conduction disease result in loss of Na+ channel function. This decreased Na+ current is expected to slow the rise time of the cardiac action potential and decrease the depolarising current to neighbouring myocytes, thereby slowing cardiac conduction. The ultimate phenotypic manifestation of reduced Na+ current (Brugada syndrome and/or conduction disease) is likely the result of a complex interplay between yet unknown factors.
Linkage to chromosome 19q13.2–q13.3 and chromosome 16q23–24 has respectively been identified in two kindreds with the disorder (table 1). The causative gene at each of these loci is unidentified.
IDIOPATHIC SICK SINUS SYNDROME
Idiopathic (congenital) sick sinus syndrome refers to the occurrence of sinus node dysfunction in the absence of identifiable acquired cardiac conditions such as ischaemic heart disease, cardiomyopathy, congestive heart failure, or metabolic diseases. It is characterised by inappropriate sinus bradycardia, sinus arrest, or chronotropic incompetence.
Mutations in HCN4 have been described for two sporadic cases with the disorder (table 1). HCN4 encodes a channel which conducts one of the currents underlying the slow diastolic depolarisation of pacemaker cells. One of the HCN4 mutations leads to a truncated channel protein lacking the domain for cAMP binding, which disrupts the cAMP regulation of the channel, explaining the chronotropic incompetence during exercise observed in this patient. The other mutation involved an amino acid substitution which led to a reduced membrane expression of the channel.
Although the role of the Na+ channel encoded by SCN5A in sinoatrial node depolarisation is unclear, compound SCN5A mutations have been identified in five patients from three families with sinus node dysfunction. The phenotype in these individuals comprised bradycardia that progressed to atrial inexcitability during the first decade of life, as well as prolonged QRS intervals and delayed His ventricle conduction.
CATECHOLAMINERGIC POLYMORPHIC VENTRICULAR TACHYCARDIA
Arrhythmias in the setting of catecholaminergic polymorphic ventricular tachycardia (CPVT) are typically bidirectional and polymorphic VT exclusively triggered by adrenergic stimuli. The phenotype often presents in early childhood but asymptomatic patients until their mid 30s have been described. In the majority of cases, CPVT displays an autosomal dominant mode of inheritance and is caused by a mutation in the gene encoding the ryanodine receptor channel (RYR2) (table 1, fig 1). This is an intracellular Ca2+ release channel on the sarcoplasmic reticulum (SR) that releases Ca2+ in response to Ca2+ entry through the membrane L-type Ca2+ channels during the action potential plateau. A recessive form of CPVT has also been recognised. This is caused by homozygous mutation in the CASQ2 gene (table 1), which encodes calsequestrin, a protein that serves as the major Ca2+ reservoir within the lumen of the SR. Symptoms are apparently more severe in CASQ2 related CPVT, including an earlier age of onset. Furthermore, diagnosis is more difficult because of the absence of a positive family history, due to the recessive nature of the disease.
Spontaneous SR Ca2+ release as a consequence of Ca2+ overload in the SR during diastole causes a depolarising transient inward current leading to deflections in the sarcolemmal membrane potential known as delayed after depolarisations (DADs). If DADs have amplitude greater than the threshold potential, depolarisation will occur, and an arrhythmia can be triggered. DAD mediated triggered activity is believed to be the principal mechanism for CPVT associated exercise induced arrhythmias. Although the precise pathophysiological mechanism(s) remains to be resolved, an enhanced SR Ca2+ release appears to underlie the pathogenesis of CPVT causing RYR2 and CASQ2 mutations.11
ATRIAL FIBRILLATION
Atrial fibrillation is the most frequent arrhythmia and occurs in particular at older age in the presence of structural heart disease. However, familial forms of atrial fibrillation occurring in the structurally normal heart, known as lone atrial fibrillation, have long been recognised. Recently two genes and two chromosomal loci, containing yet unknown genes (table 1), have been identified. A mutation in the KCNQ1 gene was identified in a large kindred with the disorder. The other gene thought to be involved is KCNE2, with mutations being identified in two probands. However, since some family members carrying the gene defect did not have atrial fibrillation, the significance of KCNE2 mutation in causality of atrial fibrillation awaits further studies. For both genes, mutation results in gain of channel function. Potentially, the genetic dissection of lone atrial fibrillation will enable research into new therapeutic options for the management of the more common forms of atrial fibrillation.
CLINICAL IMPLICATIONS OF IDENTIFICATION OF GENETIC DEFECTS IN THESE DISORDERS
The classification of the long QT syndromes into various genetic classes has led to a number of genotype–phenotype studies that have demonstrated that the underlying genetic defect impacts on ECG morphology, trigger and onset of symptoms, prognosis and, most importantly, treatment. In fact, soon after the recognition of different genetic subtypes, typical ECG features were described for LQT1, 2, and 3.12 Adrenergic stimuli proved to be the most prevalent triggers in LQT1,13 in addition to diving and swimming which are almost exclusive to this patient group.w38 w39 Patients with LQT3 are at particularly high risk at rest or during sleep since their QT interval is prolonged excessively at slow heart rates but shortens adequately during increase in rate. LQT2 patients share aspects of both LQT1 and LQT3—that is, they tend to suffer events both at rest and during exercise. Auditory stimuli, such as an alarm clock or a ringing telephone, occur almost exclusively in patients with LQT2.w38 The age of onset seems also to depend on the gene involved. At age 10 almost 40% of LQT1 children have become symptomatic, whereas only 10% of LQT2 and hardly any LQT3 patients have symptoms.14
These gene specific features form the rationale for tailored therapeutic management of genotyped LQT patients. Lifestyle adjustments to avoid genotype specific triggers are pertinent. In concordance with the predominant adrenergic triggers, β blocking treatment is most effective in LQT1 and to some extent in LQT2.13,15 In LQT3 patients β blockers seem of no use. In individual patients/families, Na+ channel blockers have been used successfully to shorten the QT interval.w40 It is important to note that long term efficacy studies with this treatment are not available. Pacing might be important in this latter patient group to avoid bradycardia related tachyarrhythmia.w41 For both these treatment strategies however, failures have been described, such that an implantable cardioverter-defibrillator (ICD) as a backup treatment appears a must. The genotype specific age of onset of symptoms has as yet not led to variable onset of treatment. Besides this, the identification of the molecular basis for these diseases opens new avenues for research into new forms of treatment for the future (for example, therapy directly opposing the molecular defect, or gene therapy).
For all arrhythmia syndromes the identification of the mutation within an affected family allows diagnosis in other family members independently from the ECG features and the arrhythmic manifestations. As for many other mendelian disorders, reduced penetrance and variable expression are more rule than exception. Hence, not all mutation carriers are clinically affected to the same degree by the disorder. Thus, extensive phenotypic variability is observed among family members carrying an identical mutation in a single ion channel gene, with far reaching implications for diagnosis and treatment. Clinical examples are particularly observed in kindreds with LQTS or Brugada syndrome where some individuals carrying the mutation may exhibit overt ECG abnormalities or suffer fatal arrhythmias, while others carrying the same primary genetic mutation might not have the ECG changes or may never develop any arrhythmias. In general, the more prominent the phenotype the higher the risk for affected patients, but data from LQTS patients show that also silent mutation carriers (that is, with QT intervals within the normal range) are susceptible to develop arrhythmias. Moreover, these individuals have a 50% chance of transmitting the genetic defect to their offspring, who in turn might be symptomatic at an earlier age. Hence, the identification of such silent mutation carriers is important.
Within the field of cardiac arrhythmias genetic aberrancies, in particular in the cardiac Na+ channel, have been observed to cause a variety of phenotypes (pleiotropy) including QT prolongation (LQT3), right precordial ST elevation (Brugada syndrome), conduction disease at various levels within the heart, sinus node dysfunction, and even dilated cardiomyopathy (table 1).16 More and more families are described with different combinations of these features. The first example of such an “overlap syndrome” is the large family that we described in 1999 in which a malignant phenotype was associated with generalised conduction abnormalities, bradycardia dependent QT prolongation, ST segment elevation in V1–V3, and bradyarrhythmias partly based on intrinsic sinus node dysfunction.17 Extensive studies on the biophysical properties of the mutant channels revealed a satisfactory explanation for the diversity in phenotype.w42 w43 However, this pleiotropy also provides evidence that genetic modifiers play an important role in determining the ultimate phenotype and severity of disease in cardiac channelopathies.
Genetics of primary cardiac arrhythmia syndromes: key points
-
Several cardiac arrhythmia syndromes that for long were considered idiopathic are now known to have a genetic basis. They are caused by mutations in genes primarily encoding ion channels
-
The genetic basis for most arrhythmia syndromes is heterogeneous—that is, a given disorder may be caused by mutations in different genes, and evidence for further genetic heterogeneity exists
-
Mutations in cardiac ion channels lead to abnormal ionic current characteristics via mechanisms such as defective channel gating or reduction in sarcolemmal channel expression. This leads to the ECG features and/or arrhythmogenesis in the inherited arrhythmia syndromes
-
It is becoming increasingly clear that treatment should take the type of gene affected into consideration (gene specific therapy)
-
The identification of a proband with a primary cardiac arrhythmia should trigger screening of family members to identify all affected relatives (presymptomatically)
-
In the long QT syndrome, information on ECG morphology and triggers for arrhythmia (to be sought in the proband as well as family members) can indicate the gene likely affected, enabling initiation of the most appropriate treatment even before results from the genetic test become available
-
Considerable heterogeneity may exist in disease manifestation (both in severity as well as differences in disease features) among family members carrying the same mutation. In this respect, a genetic test is vital in uncovering all carriers of the genetic defect within a family
A good example of the importance of modifier genes is a family described with the rare arrhythmia familial atrial standstill.18 In this family a Na+ channel mutation with mild biophysical effects (D1275N) co-segregated with a discrete “conduction phenotype”. However, in the presence of homozygosity for two closely linked single nucleotide polymorphisms (SNPs) in the promoter region of the connexin 40 gene (Cx40), which are potentially associated with reduced expression of Cx40, atrial standstill was observed. Hence, the SCN5A mutation leads to an atrial phenotype only in the presence of the Cx40 modifier which exclusively acts at the level of the atria. Of note, in another family the same SCN5A mutation was associated with dilated cardiomyopathy, in particular when associated with atrial arrhythmias.16 The concept that ion channel disorders can lead to structural heart disease is of great interest. In a previously described small nuclear family, compound heterozygosity for two SCN5A mutations was associated with severe conduction disease and proven fibrosis in the conduction system.19 In the years to come we will certainly learn more about this intriguing association between electrical and structural heart disease.
FUTURE RESEARCH
While the identification of genes associated with primary electrical disease has progressed at a rapid momentum, insight into the genetics of “acquired” arrhythmias such as in ischaemic heart disease, hypertrophy, heart failure or during the use of (QT interval prolonging) medications, is still in its infancy and examples of such studies are very sparse in the literature. The identification of susceptibility genes for acquired arrhythmias, together with the identification of genetic modifiers of primary electrical disease, actually represents an anticipated challenging next step in our understanding of the genetics of arrhythmias. Here, polymorphisms—that is, common variants that are present (in relevant genes) throughout the genome—are expected to influence the susceptibility to arrhythmias. Evidence for such an effect has been provided by the finding that a variant leading to an amino acid substitution in SCN5A (S1103Y), found primarily in individuals of African descent (and absent in whites), was more prevalent in individuals being treated for arrhythmia than in the general population, suggesting that it increases risk.20
The identification of such susceptibility genes for acquired arrhythmias necessitates the construction of large databases of patients phenotyped in a very standardised fashion to ensure adequate power for association studies. A large number of genes are expected to contribute to susceptibility. Polymorphisms in genes encoding cardiac ion channels form very plausible candidates in which to search, but then the multitude of other players within the diverse pathways leading to arrhythmia, such as cytoskeleton proteins, structural proteins (such as those linked to sudden death in familial hypertrophic cardiomyopathy) and proteins involved in processes such as fibrosis, inflammation, cell-to-cell communication, and electrical and structural remodelling, are also of interest.w44 With recent advances in genotyping technology, comprehensive screening of multiple genes in different pathways is now feasible. Moreover, the availability of a high resolution genome-wide map of polymorphisms and the evolving technology for large scale genome-wide screens (unbiased approach), and related bioinformatics, are also expected to facilitate the study of genetics of common acquired arrhythmias. This is likely to provide novel tools for risk stratification and open new opportunities for prevention and therapy of lethal arrhythmias in the common pathologies.
Additional references appear on the Heart website—http://www.heartjnl.com/supplemental
Acknowledgments
Work from the author’s laboratory is funded by Netherlands Heart Foundation grants: 2000B059, 2003B195 and 2003T302 and ICIN (project 27).
REFERENCES
Supplementary materials
The references are available as a downloadable PDF (printer friendly file).
If you do not have Adobe Reader installed on your computer,
you can download this free-of-charge, please Click hereFiles in this Data Supplement:
- [view PDF] - Web-only references.
Footnotes
-
In compliance with EBAC/EACCME guidelines, all authors participating in Education in Heart have disclosed conflicts of interest that might cause a bias in the article