Article Text

Statistics from Altmetric.com
It was once thought that aldosterone was a hormone released purely from the adrenal cortex, which circulated in the bloodstream and acted only on the kidneys to retain sodium and excrete potassium. This is now recognised to be a gross oversimplification (table 1).
The new biology of aldosterone
NEW BIOLOGY OF ALDOSTERONE
The first novel finding discovered about aldosterone is that it is made locally in various different tissues in the body including the brain, vascular tissue, and the myocardium.1,2 In heart failure, myocardial tissue synthesises even more aldosterone. The second novel finding is that mineralocorticoid receptors which are activated by aldosterone are in fact also widespread in the body including the brain, vascular tissue, and the myocardium. This means that aldosterone may act in a paracrine fashion in many tissues—that is, locally made aldosterone may act on local aldosterone receptors to mediate local (mostly adverse) effects. This may be one of many reasons why baseline plasma aldosterone values appear to only poorly predict the efficacy of aldosterone blockade. Other reasons may also explain this (see below and table 2).
Reasons why baseline plasma aldosterone concentrations may not predict the response to aldosterone blockade
However, the main revolution in our new understanding of aldosterone is that it has now been shown to mediate a host of different adverse biological effects in the body, which have only been recognised in the last 10 years. These effects range from vascular endothelial dysfunction to inflammation to widespread tissue injury and repair. These were often first seen in animal models and then found also in man.
In vitro studies showed first that aldosterone reduced nitric oxide produced in response in inflammatory stimuli. In experimental animals in vivo, aldosterone was then found to produce a vascular inflammatory response which is characterised not only by increased expression of cytokines such as osteopontin, but also by tissue injury in several different organs (myocardium, brain, and kidney). In various different animal models, Rocha and colleagues3 have shown that aldosterone blockade reduces both tissue injury and tissue fibrosis in the myocardium, in the kidney, and in the brain. This tissue protection is seen even when aldosterone blockade is given at a dose too low to alter blood pressure—that is, the tissue protective effect of aldosterone blockade in experimental models is not simply due to its antihypertensive effect. A similar effect may also occur in man (see below) but more data are required before it can be fully accepted that aldosterone blockade specifically reduces tissue injury in man.
The vascular effects of aldosterone may be due to aldosterone increasing free radicals which then inactivate nitric oxide (NO). Such an effect on free radicals could be due to aldosterone increasing NAD(P)H oxidase activity which normally generates superoxide anions. In animal models of atherosclerosis, a similar effect is seen in that the specific aldosterone blocker, eplerenone, decreases NADH/NADPH oxidase dependent free radical production.4 Extending these observations further is a study showing that spironolactone reduces the p22phox subunit of NADH oxidase. Thus aldosterone may reduce NO bioactivity by increasing NADH oxidase induced free radical production which in turn degrades NO.
Another major effect of aldosterone in experimental animals is to produce tissue fibrosis.5 Intriguingly, in animal models, aldosterone only produces myocardial fibrosis when it is administered along with a high salt diet. How relevant a high salt diet is to aldosterone induced tissue damage in man is completely unknown, but salt status could be a key determinant of whether aldosterone produces tissue damage in man. Patients with heart failure all tend to have increased total body sodium as well as increased aldosterone concentrations and hence they resemble the animals studied by Weber5 who received both aldosterone and a high salt diet.
UNRESOLVED INTRIGUING MECHANISTIC ISSUES
The biology of aldosterone has turned out to be amazingly complex and many surprising features have arisen with regard to the new biology of aldosterone. Scientists are trying to shed further light on these complex issues. In order for the clinician to gain some insight into these complex issues, I will give a brief overview of a couple of the intriguing issues currently taxing basic scientists involved in aldosterone research. Each of these basic issues may have clinical implications.
The traditional mineralocorticoid receptor appears not to mediate all the effects of aldosterone. Aldosterone stimulation of the traditional mineralocorticoid receptor in the cytoplasm exerts its biological effect slowly (after 1–2 hours) by way of stimulating transcription, translation, and the expression of new proteins. In addition to this traditional route, aldosterone has been shown to exert some rapid effects which are considered non-genomic because they occur within minutes and within a timescale which could not possibly involve transcription or translation. The intriguing thing about these fast, non-genomic effects of aldosterone are that they are not exerted through the traditional mineralocorticoid receptor and hence drugs such as spironolactone do not prevent them. It is currently unclear which biological effects of aldosterone occur by the traditional mineralocorticoid receptor and which occur by the fast, non-genomic route. Most of the effects described below do appear to occur by the former traditional route.
The second unusual feature about aldosterone biology is that the traditional mineralocorticoid receptor is actually stimulated equally by glucocorticoids (cortisol) and mineralocorticoids (aldosterone). The added twist which makes this intriguing is that plasma cortisol concentrations are generally about 10 times as high as plasma aldosterone concentrations. This means that one would expect the traditional mineralocorticoid receptor to be stimulated more in vivo by cortisol than by aldosterone. What prevents this happening is a bizarre arrangement whereby an enzyme called 11β hydroxysteroid dehydrogenase (11 βHSD) sits close to the mineralocorticoid receptor and inactivates cortisol before it can stimulate the mineralocorticoid receptor. This enzyme does not inactivate aldosterone, which means that the mineralocorticoid receptor is in fact mainly stimulated in vivo by aldosterone because this enzyme inactivates cortisol which tries to bind to the mineralocorticoid receptor. Not only is this arrangement bizarre, but it could be of clinical relevance. Some diseases might alter activity of 11 βHSD and hence alter the normal selectivity of the mineralocorticoid receptor for mineralocorticoids over glycocorticoids. For example, in experimental diabetes, the activity of this enzyme in the kidneys is reduced which could lead to more stimulation of the mineralocorticoid receptor by glucocorticoids. The other possible clinical consequence could be that plasma aldosterone values may not be any indication of the efficacy of aldosterone blockade because the mineralocorticoid receptor can be occupied and activated also by cortisol in certain circumstances (table 2). Indeed the fact that eplerenone reduces blood pressure equally in all types of hypertension (low renin and high renin) may be an example of this very fact (although other explanations are possible) (table 2).
NEW BIOLOGY OF ALDOSTERONE (CLINICAL ASPECTS)
An intense clinical research effort has also revealed a host of new adverse effects produced by aldosterone (table 1). These in general support most of the experimental data described above.
Endothelial dysfunction
The endothelium plays a critical role in regulation of vascular tone, platelet aggregation, adhesion of leucocytes, and thrombosis. A growing body of evidence suggests that aldosterone can cause endothelial dysfunction which then makes the vessel “sticky” and prone to microthrombi. Because of this endothelial dysfunction is now recognised as an excellent predictor of future cardiovascular events.6
The clearest indication of this is that in normal human volunteers an infusion of aldosterone which does not alter blood pressure does in fact produce endothelial dysfunction.7 This phenomenon has been called “aldosterone induced vasculopathy”.
Before this, the first suggestion that aldosterone induced vasculopathy occurred in man had come from a study we did in chronic heart failure (CHF) patients which showed that aldosterone blockade with spironolactone increased vascular NO bioactivity.8 In this study infusion of the NO synthase inhibitor L-NMMA resulted in significantly greater vasoconstriction in spironolactone treated patients compared to those in the placebo group, indicating that basal NO bioactivity had been increased by aldosterone blockade. In the same study, spironolactone was also associated with a significant increase in forearm blood flow in response to acetylcholine, but had no effect on blood flow in response to sodium nitroprusside, an endothelium independent vasodilator. These are exactly the findings one expects to see when a treatment (aldosterone blockade) improves endothelial function by increasing vascular NO bioactivity.
As mentioned above, in animal models, aldosterone induced vasculopathy has an inflammatory element—that is, it is more like an aldosterone induced vasculitis. However, there is as yet no firm evidence of aldosterone being pro-inflammatory in man. Indeed, we have assessed the effect of spironolactone on plasma concentrations of C reactive protein (CRP) (high sensitivity) in heart failure and found that spironolactone did not alter CRP values.
Myocardial fibrosis and cardiac remodelling
Aldosterone also contributes to the progression of heart failure by promoting perivascular and interstitial myocardial fibrosis. This has several different but important consequences. Firstly, it reduces the flexibility of myocardial tissue and could cause “diastolic dysfunction”. Secondly, patchy myocardial fibrosis would also be expected to produce electrical inhomogeneity which would be arrhythmogenic. Such arrhythmogenicity may be further potentiated by the potassium and magnesium depletion induced by aldosterone. Illustrative of this is that ventricular ectopy on 24 hour ambulatory ECG falls when aldosterone blockade is administered.9
The pro-fibrotic effects of aldosterone have been demonstrated at a cellular level, preclinically, and in several clinical settings. The first proof that aldosterone promotes myocardial fibrosis in man came when it was shown that spironolactone reduced plasma concentration of PIIINP in heart failure.10 PIIINP is procollagen type III amino terminal peptide and is an indirect marker of myocardial collagen turnover in man. A similar effect of spironolactone in reducing PIIINP was also seen in the RALES study.11 Indeed, the antifibrotic effect of spironolactone may partially explain the RALES result, since spironolactone only reduced mortality in RALES in those patients who had an above normal concentration of PIIINP.
The adverse effects of aldosterone on endothelial function could account partially for its pro-fibrotic action. Endothelial dysfunction could lead to microthrombi, tissue injury, and tissue microinfarction, which repairs itself as fibrosis. Whether aldosterone produces fibrosis directly or whether it acts via a vasculopathy induced injury of tissues is an intriguing and as yet unanswered question. A related adverse effect of aldosterone is that it produces adverse left ventricular (LV) remodelling. Spironolactone has been shown clearly to reduce LV dilatation and improve the LV ejection fraction.12 These effects should improve exercise capacity and reduce deaths caused by progressive heart failure.
Another potentially harmful effect of aldosterone is its ability to blunt the baroreflex response. Perfusing the carotid sinus directly with aldosterone has been shown to reduce maximum baroreceptor discharge, while in dogs chronic administration of aldosterone elevates the threshold for baroreflex activation and decreased peak discharge rate. The first demonstration of such an effect in man came when it was shown that aldosterone inhibits baroreflex sensitivity in normal human volunteers.13
HEART FAILURE
The main indication now for aldosterone blockade is heart failure. Like many neurohormones, plasma aldosterone concentrations are increased in heart failure. However, the advent of angiotensin converting enzyme (ACE) inhibitor treatment for congestive heart failure, led initially to the feeling that any adverse effects of aldosterone would be ameliorated by ACE inhibitors. It is, however, now apparent that although ACE inhibitors produce an acute fall in plasma aldosterone, the concentration of aldosterone gradually rises again, and indeed returns to baseline or higher in some patients: this phenomenon has been called “aldosterone escape”.
Trial acronyms
-
EPHESUS: Eplerenone Part Acute Myocardial Infarction Heart Failure Efficacy and Survival Study
-
HOPE: Heart Outcomes Prevention Evaluation
-
RALES: Randomized Aldactone Evaluation Study
-
RESOLVD: Randomized Evaluation of Strategies for Left Ventricular Dysfunction
Aldosterone concentrations > 144 pg/ml were reported to occur in up to 40% of patients with symptomatic CHF14 despite use of an ACE inhibitor. In another study, six weeks of captopril produces only a mean fall of 20% in plasma aldosterone concentrations. In this latter study, the aldosterone values varied greatly between patients, from 56 pmol/l to 1568 pmol/l, despite captopril treatment.
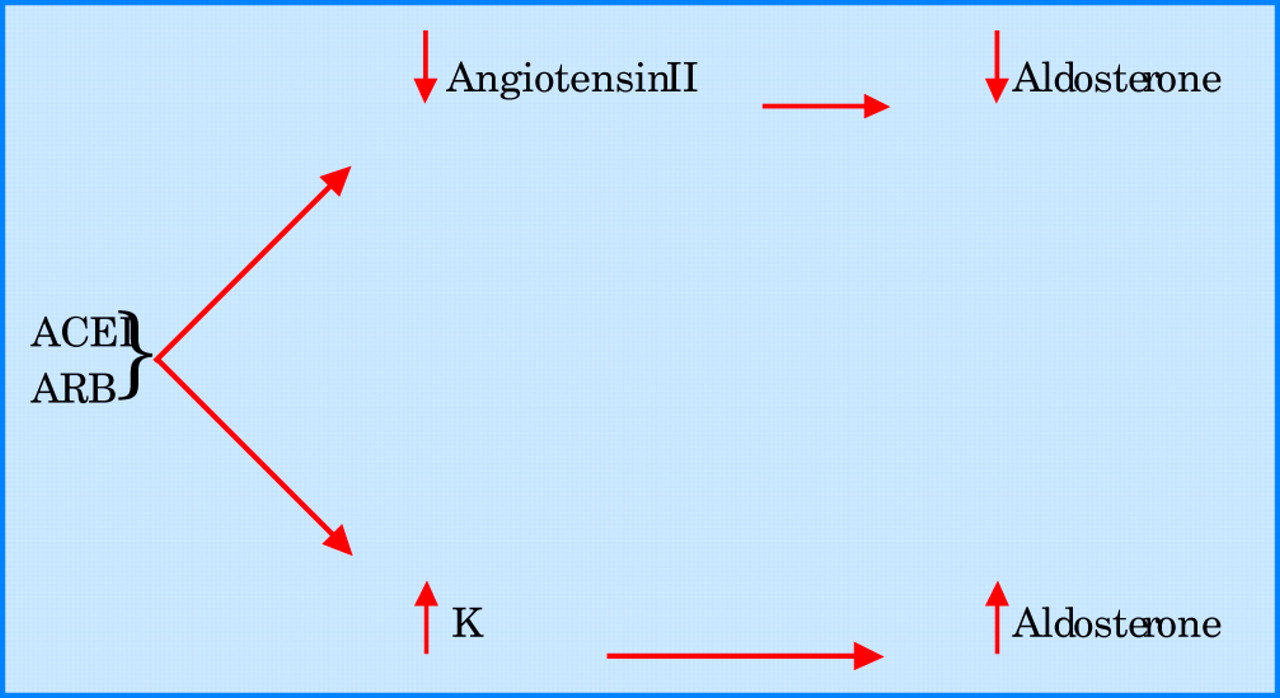
Even when an ACE inhibitor is given in combination with an angiotensin I receptor antagonist, aldosterone concentrations remain uncontrolled. In the RESOLVD pilot study, patients with CHF who were given both enalapril and candesartan had a significant fall in aldosterone at 17 weeks, but mean aldosterone concentrations had returned to baseline by 43 weeks even with maximum doses of both agents. The precise mechanism by which aldosterone values rise during ACE treatment is unclear. However, it is worth noting that angiotensin II is only one of many secretagogues for aldosterone. Another key secretagogue is potassium. Obviously ACE inhibitors increase potassium, and since potassium is a powerful secretagogue for aldosterone, this may be a major reason for aldosterone escape (fig 1). This means that it is probably impossible to neutralise aldosterone completely by blocking angiotensin II, because blocking angiotensin II is inevitably accompanied by a potassium increase which then increases aldosterone.

Mechanisms of aldosterone escape with angiotensin converting enzyme inhibitor (ACEI) or angiotensin receptor blocker (ARB) treatment. K, potassium.
The main demonstration of aldosterone’s harmful effects comes from two major prospective trials. In the RALES study, patients with severe CHF (New York Heart Association (NYHF) functional class III–IV) were randomised to receive spironolactone or placebo.15 The trial was discontinued early because after a mean follow up of 24 months, the relative risk (RR) of death was 0.70 (95% confidence interval (CI) 0.6 to 0.82; p < 0.001) among patients receiving spironolactone—that is, a 30% reduction in risk of death with aldosterone blockade. This reduction in mortality was accounted for by both a significant fall in deaths caused by progression of heart failure (RR 0.64, 95% CI 0.51 to 0.80; p < 0.001) and to sudden cardiovascular death (RR 0.71, 95% CI 0.54 to 0.95; p = 0.02). However, gynaecomastia or breast pain occurred more often in men receiving spironolactone than placebo (10% v 1%; p < 0.001) due to the drug’s affinity for androgen receptors. The low dose of spironolactone used in the RALES study is said to have no apparent diuretic effect, as judged by a substudy where the sodium retention score was measured.
The reduction in sudden cardiovascular death seen in RALES could be due to many possible mechanisms, ranging from aldosterone worsening endothelial dysfunction and so increasing acute coronary events, to it having arrhythmogenic effects by promoting myocardial fibrosis and depleting potassium and magnesium.9 These mechanisms are illustrated in fig 2. The reduction in progressive heart failure deaths was presumably caused by spironolactone improving LV remodelling (see above).

{kind=link}
{kind=link}
Mechanisms whereby aldosterone promotes sudden cardiac death.
More recently, the EPHESUS trial has evaluated use of the selective aldosterone blocker eplerenone in 6632 patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure.16 Severity of CHF was less pronounced than in RALES, with mean left ventricular ejection fraction of 33% compared to 25% in the RALES population. Pharmacotherapy also differed: most notably, 75% of patients received β blockers versus approximately only 10% of those in RALES. During a mean follow up of 16 months, patients randomised to eplerenone had a 15% reduction in mortality compared to patients on placebo, and risk of hospitalisation for heart failure also fell by 15%. Similar to the RALES study, there was a large fall (21% fall) in sudden cardiac death. This indicates that the myocardial protective effect of aldosterone blockade is maintained even in the presence of optimal treatment and in patients close to the acute phase of myocardial infarction. Incidence of gynaecomastia and impotence did not differ between the eplerenone and placebo groups, due to the much lower affinity of eplerenone for androgen receptors.
HYPERTENSION
The other main indication for aldosterone blockade is hypertension. Several published trials show that the selective aldosterone antagonist, eplerenone, reduces blood pressure in hypertension. The first point of note is that eplerenone appears to reduce blood pressure equally in all different subgroups of hypertension.17 Eplerenone also reduces blood pressure effectively when given in addition to an ACE inhibitor or angiotensin receptor blocker (ARB) treatment.18 The eplerenone induced reduction in systolic blood pressure was 13.4 mm Hg in those patients on ACE inhibitors and −16 mm Hg in those on ARB treatment, in comparison to −8/9 mm Hg in response to placebo. In a comparison with losartan, eplerenone reduced blood pressure similarly in white patients and to a greater extent than losartan in black patients. The latter result is not too surprising since ARBs are generally less effective antihypertensives in blacks. Before this result, one might have thought that blacks would respond poorly to eplerenone because blacks tend to have low renin hypertension (which is why they respond poorly to angiotensin blockade). However, this was not seen here which emphasises that for some reason (see below), eplerenone appears to reduce blood pressure equally in all types of hypertension (young, old, black, white, low renin, high renin).
The second main feature about eplerenone in hypertension is that early data do indeed suggest the drug has a target organ protection effect. White and colleagues19 found in isolated systolic hypertension that eplerenone and amlodipine reduced the systolic blood pressure equally, but that eplerenone outperformed amlodipine in terms of reducing microalbuminuria. An effect of reducing microalbuminuria was also seen by Epstein and colleagues20 in diabetic hypertensives. Indeed, eplerenone added to enalapril reduced microalbuminuria more than either alone, although potassium needed to be carefully monitored when eplerenone was given together with an ACE inhibitor in diabetics with microalbuminuria. The third example of target organ protection with eplerenone was a study of hypertensives with LV hypertrophy where eplerenone and enalapril both reduced LV mass but more so when they were combined.21
In all the above hypertension studies, eplerenone was generally well tolerated. Only in diabetics with microalbuminuria given eplerenone and an ACE inhibitor was there concern about the concentrations of potassium attained. Despite this, potassium and creatinine do need to be monitored after eplerenone in all patients as increases in both can occur in some patients.
The question why eplerenone reduces blood pressure equally in all types of hypertension is an intriguing one. One possibility is that in high renin patients eplerenone reduces blood pressure by acting as a neurohormonal antagonist, while in low renin patients a subtle natriuretic effect of eplerenone is the cause of the blood pressure reduction.
Interestingly hypertension experts tend to be divided as to whether the first antihypertensive drug to be used should be a thiazide or an ACE inhibitor. Eplerenone may act as a bit of both—a diuretic and a neurohormonal antagonist. This does not, however, mean that eplerenone will become a first line antihypertensive in the near future. It does mean, however, that it will be a useful add-on antihypertensive drug, producing a fairly predictable added hypotensive effect in all patients. In addition, it appears to especially protect target organs and it may be particularly beneficial to add to angiotensin inhibiting/blocking treatment in those patients with target organ damage in the form of LV hypertrophy or diabetic microalbuminuria.
OTHER CLINICAL INDICATIONS
Although heart failure and hypertension are the main clinical indications for aldosterone blockade, several other options ought to be explored in the future (table 3). There are good experimental data that aldosterone blockade reduces tissue injury in the myocardium, the kidney, and the brain. There are promising signs from clinical studies that aldosterone blockade may protect target organs, especially left ventricular hypertrophy and microalbuminuria. This suggests the possibility that aldosterone blockade may slow down the deterioration in renal function seen during progressive renal disease. This may occur over and above traditional ACE inhibition in this setting. The other major possibility is that aldosterone blockade will prevent future cardiovascular events in the type of vascular patients seen in the HOPE and EUROPA studies. Again such an effect may occur on top of traditional ACE inhibitor treatment.
Clinical indications for aldosterone blockade
ADVERSE EFFECTS OF ALDOSTERONE BLOCKADE
The adverse effects of eplerenone and spironolactone may result in an increase in serum potassium concentrations. In clinical trials, the adverse effect profile of eplerenone given along or in combination with other antihypertensive medications was not significantly different from that of placebo, with the exception of this increased risk of hyperkalaemia. In EPHESUS, the incidence of serious hyperkalaemia, defined as a serum potassium concentration ⩾ 6 mEq/l, was 5.5% in the eplerenone group and 3.9% in the placebo group (p = 0.002). In EPHESUS, this was counterbalanced by eplerenone reducing the incidence of hypokalaemia (13% to 8%).
The increased incidence of hyperkalaemia with eplerenone is similar to that seen with spironolactone. In RALES, the incidence of serious hyperkalaemia, defined as a serum potassium concentration > 5.5 mEq/l, increased with increasing dosages of spironolactone, from 5% with 12.5 mg/day to 13% with 25 mg/day, 20% with 50 mg/day, and 24% with 75 mg/day. As expected, the rate of sex hormone related adverse events has been much lower with eplerenone than with spironolactone. The incidence of gynaecomastia or breast pain was significantly greater in men receiving spironolactone compared with those receiving placebo (10% v 1%, respectively; p < 0.001). With eplerenone, on the other hand, sex hormone related events have been reported to be no greater than that seen with placebo.
The US Food and Drug Administration (FDA) have licensed eplerenone for use in hypertension but coincidental diabetic microalbuminuria was recommended by them to be a specific area for concern. This was because the incidence of hyperkalaemia was higher in this subgroup. To some extent, this was an odd decision by the FDA because eplerenone was particularly efficacious in this subgroup in terms of reducing microalbuminuria. However in this subgroup, the greater efficacy of eplerenone was indeed matched by a greater incidence of the adverse effect of hyperkalaemia. This is a good example of how in clinical therapeutics, benefit and risk sometimes go together. It might mean that in the future, a careful monitoring schedule could be devised so that in this subgroup of patients, the greater efficacy of eplerenone could be harnessed while also identifying those individuals who develop hyperkalaemia so that treatment in those individuals could be terminated.
CONCLUSIONS
A range of newly recognised harmful effects of aldosterone may contribute to its detrimental effect on cardiovascular events, in particular on sudden death. Apart from the well recognised effect of potassium and magnesium depletion, studies have shown that aldosterone promotes endothelial dysfunction and cardiac fibrosis, actions which would be expected to play an important part in promoting cardiac events, cardiac arrhythmias, and cardiac death.
We now have convincing clinical evidence that these adverse effects of aldosterone translate into increased mortality in patients with CHF. Two large scale prospective studies have reported a pronounced and significant benefit from use of an aldosterone blocker in terms of overall mortality, cardiovascular mortality (particularly sudden death), and hospitalisation. In one trial, these improvements were seen even when patients were already taking an ACE inhibitor and a β blocker in addition to aldosterone blockade. Optimal treatment for patients with congestive heart failure should now routinely include an aldosterone blocker since two major trials clearly documented a highly significant reduction in total mortality by doing so. The only remaining question is how early in the heart failure disease process should we initiate aldosterone blockade.
Aldosterone blockade also reduces blood pressure in virtually all patients with hypertension and it is likely that eplerenone will become a very useful second or third line antihypertensive drug. Aldosterone blockade seems especially able to protect target organs (left ventricular hypertrophy and microalbuminuria) and may prove ultimately to be particularly useful (in addition to ACE inhibitors) in patients who already have these forms of hypertensive target organ damage.
REFERENCES
Supplementary materials
Web-only References
The references are available as a downloadable PDF (printer friendly file).
If you do not have Adobe Reader installed on your computer,
you can download this free-of-charge, please Click hereFiles in this Data Supplement:
Linked Articles
- Miscellanea