Article Text

Abstract
Chronic kidney disease is associated with elevated cardiovascular risk, and heart failure and arrhythmias are the biggest causes of cardiovascular death in this population. Increased arterial stiffness is a hallmark of chronic kidney disease and is associated with adverse alterations in cardiac structure and function that may predispose to an increased risk of cardiovascular death. These changes are already apparent in early kidney disease, which is highly prevalent in the developed world. The mechanisms underlying increased arterial stiffness in chronic kidney disease are undoubtedly complex, but an understanding is paramount to enable the development of novel therapeutic strategies to prevent or reverse this pathophysiology and therefore reduce the cardiovascular disease burden in this high-risk cohort.
- Arterial stiffness
- chronic kidney disease
- heart failure
- diastolic dysfunction
- left ventricular hypertrophy
- renal disease
Statistics from Altmetric.com
- Arterial stiffness
- chronic kidney disease
- heart failure
- diastolic dysfunction
- left ventricular hypertrophy
- renal disease
The importance of arterial stiffness in chronic kidney disease
Chronic kidney disease and cardiovascular risk
Chronic kidney disease (CKD) confers an elevated risk of cardiovascular disease with an inverse graded relationship with glomerular filtration rates <60,1 and perhaps < 90 ml/min/1.73 m2,2 independent of other risk factors. Patients with CKD are far more likely to die from cardiovascular disease than progress to end-stage kidney disease (ESKD) requiring dialysis or transplantation.3 Although cardiovascular risk in ESKD is extreme, the public health burden of cardiovascular disease caused by early-stage CKD is much greater; the prevalence of CKD in the developed world is 13% and appears to be rising.4 This increased cardiovascular risk cannot be explained by ‘traditional’ risk factors, many of which are inversely associated with survival in CKD, a phenomenon attributed to reverse causality.5 Research in this area has been significantly lacking in both quality and quantity.6 This may partly reflect the lack of a consistent definition of CKD, a problem now overcome by the adoption of the K/DOQI (Kidney/Dialysis Outcome Quality Initiative) classification (table 1).7
Stages of chronic kidney disease
Although about 50% of all ESKD deaths are due to cardiovascular disease, only 18% of these are attributable to vasculo-occlusive diseases such as myocardial infarction, the remainder being attributed to sudden cardiac death, arrhythmia and congestive heart failure (CHF).8 Heart failure is a major cause of morbidity and mortality in CKD with incident rates three to four times higher than in non-CKD subjects.9 Thus there is increasing evidence that structural heart disease, leading to CHF and sudden cardiac death, rather than occlusive arterial disease, is the leading cause of cardiovascular mortality in both ESKD and early-stage CKD. Powerful evidence suggests that increased arterial stiffness is a major cause of this structural heart disease.
Vascular pathology in chronic kidney disease
There appear to be two distinct vascular pathologies occurring in patients with CKD: atherosclerosis and arteriosclerosis.10 Atherosclerosis is an intimal disease characterised by fibro-atheromatous plaques and vascular occlusion. Atherosclerotic lesions in CKD are characterised by increased plaque calcification and increased intimal and medial thickness.11 Arteriosclerosis is a disease of the arterial medial layer. Increased collagen content, calcification and hyperplasia and hypertrophy of vascular smooth muscle cells (VSMC) all result in arterial wall hypertrophy and increased stiffness.10 Although associations have also been established between the degree of arterial stiffness and atheromatous plaque burden,12 recent studies have failed to demonstrate a significant influence of traditional atherosclerotic risk factors on the development of arteriosclerosis,13 suggesting that alternative pathologies are driving this process. Although endothelial dysfunction and intimal disease are known to contribute to arterial stiffening, the relationship between arteriosclerosis and atheromatous disease remains poorly understood.
The clinical importance of arterial stiffness
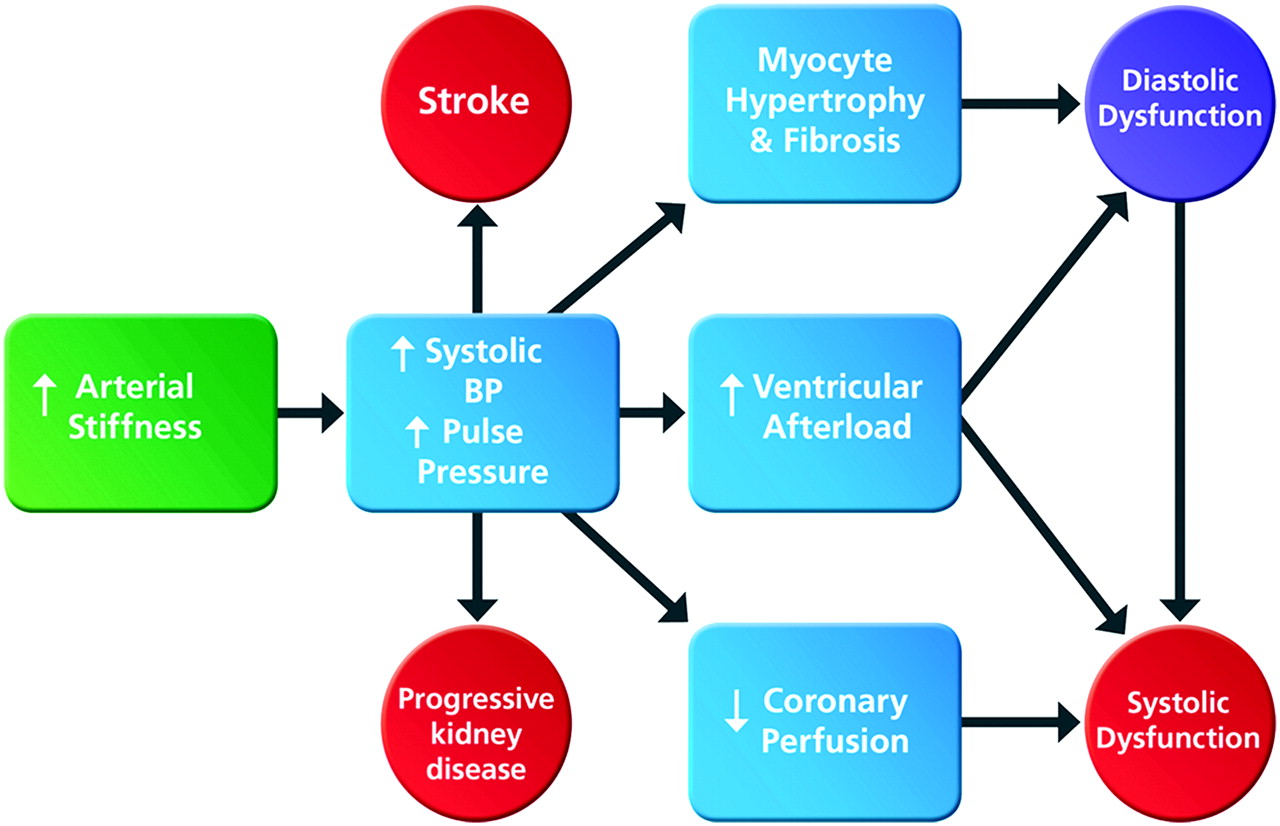
A major function of the aorta and large arteries is to buffer oscillatory changes in blood pressure that result from intermittent ventricular ejection. The highly distensible arterial system ensures that most tissues receive near steady flow with no exposure to peak systolic pressures; this mechanism is so efficient that there is almost no drop in diastolic pressure from the ascending aorta to the peripheral arteries.14 Loss of arterial distensibility results in a more rigid aorta that is less able to accommodate the volume of blood ejected by the left ventricle, resulting in greater pressure augmentation in systole and higher pulse pressures.15 An often cited explanation for the elevated systolic pressure that accompanies an increase in arterial stiffness16 is the more rapid return of reflected waves of ventricular contraction from the distal vasculature. According to this hypothesis, in healthy compliant arteries reflected waves return to the ascending aorta in diastole, thus augmenting diastolic pressure and coronary blood flow, while in aged stiffer arteries the reflected waves return earlier in systole, augmenting systolic and pulse pressures and increasing ventricular afterload. Although an attractive concept, there is now abundant evidence, recently published as a meta-analysis, showing that reflected waves arrive in systole irrespective of age.17 These data, together with information available from techniques that separate reflected waves from reservoir pressure, strongly suggest that the ‘cushioning effect’ or Windkessel model appears to be the more important physiological explanation.18 Regardless of the underlying mechanism, as arterial stiffness increases, the myocardium, brain and kidneys are exposed to higher systolic pressures and greater pressure fluctuations resulting in myocardial hypertrophy and fibrosis, cerebral and renal microvascular damage and an increased risk of stroke and renal impairment (figure 1).19 Furthermore, lower diastolic pressure reduces diastolic coronary perfusion and promotes subendocardial ischaemia and ventricular stiffening20; it also places greater reliance on systolic coronary perfusion, conferring heightened vulnerability of the myocardium to any decline in systolic function.21

Consequences of increased arterial stiffness in chronic kidney disease. Increased arterial stiffness leads to increases in systolic blood pressure (BP) and pulse pressure causing myocyte hypertrophy, increased ventricular afterload and reduced coronary perfusion, resulting in diastolic and systolic dysfunction and ultimately congestive heart failure. Raised systolic and pulse pressures also promote further vascular damage increasing the risk of stroke and further loss of kidney function.
Cardiac function is physiologically matched with arterial function through ventricular–arterial coupling to ensure maximum cardiac work and efficiency.22 When arterial stiffness increases, the left ventricle generates greater end-systolic pressures that enhance ventricular systolic wall stress and increase ventricular stiffness. These compensatory adaptations maintain cardiac performance with enhanced contractility at rest, but at a price: cardiac reserve is reduced, diastolic function is impaired and the cardiovascular response to alterations in pressure and volume load is blunted leading to haemodynamic instability.22 Myocardial oxygen consumption also increases, promoting subendocardial ischaemia.23 Increased ventricular stiffening further impairs diastolic coronary perfusion through increased compression of the coronary microvasculature.24 While this is a common feature of advanced disease characterised by left ventricular hypertrophy (LVH), it is often present despite normal ventricular wall thickness.22 Reduced aortic distensibility also correlates with reduced exercise capacity despite normal or enhanced left ventricular systolic function.25 Increased arterial stiffness due to CKD may therefore underlie many cases of heart failure with preserved ejection fraction, otherwise known as diastolic heart failure.
Ventricular function is abnormal in early chronic kidney disease
Abnormalities in left ventricular structure are evident in almost all patients starting dialysis; 70–80% have established LVH on echocardiography.26 Abnormalities of left ventricular function associated with early-stage CKD and their relationship to arterial stiffening have only recently been studied. Compared with controls, patients with stage 2 and 3 CKD had delayed ventricular relaxation, increased left ventricular end-diastolic stiffness and pressure, and elevated left atrial volumes.27 Ventricular–arterial coupling was preserved with an increase in both arterial elastance (stiffness) and left ventricular elastance, suggesting that aortic stiffness drives the development of left ventricular stiffness in early CKD.27
Left ventricular systolic dysfunction is also common in ESKD but was considered rare in early CKD. In a group of normotensive subjects with stage 2 and 3 CKD and no overt cardiovascular disease we showed that although left ventricular function was normal according to conventional echocardiographic criteria, measurements of peak systolic strain and strain rate were subnormal with evidence of impaired longitudinal tissue deformation.28 These abnormalities have been associated with adverse cardiovascular outcomes in late-stage CKD.29
Arterial stiffness and mortality
Increased arterial stiffness and systolic and pulse pressures are characteristic features of ageing in Western populations30 and are independent risk factors for mortality and development of CHF.31 Aortic pulse wave velocity (aPWV), a widely used measure of arterial stiffness, is a powerful independent predictor of all-cause mortality and cardiovascular events in ESKD, hypertensive subjects, the elderly, diabetic subjects and the general population.32
Cardiovascular risk and arterial stiffness are both increased with only minor reductions in renal function.33 Aortic distensibility measured by cardiac magnetic resonance imaging is reduced compared with controls in stage 2 and 3 non-diabetic CKD.27 An understanding of the potential mechanisms underlying increased arterial stiffness in CKD is paramount for devising strategies to prevent or reverse this pathophysiology (figure 2).

{kind=link}
{kind=link}
Potential mechanisms of increased arterial stiffness in chronic kidney disease. Alterations in the extracellular matrix and endothelial dysfunction are promoted by advanced glycation end products (AGE), activation of the renin–angiotensin–aldosterone system (RAAS), chronic inflammation and vascular calcification, all of which are hallmark features of chronic kidney disease. Vascular calcification is closely linked with chronic kidney disease–mineral bone disorder (CKD–MBD) and raised serum phosphate (PO4). Several of these features typify the ageing process but appear to be accelerated in patients with chronic kidney disease. They are also influenced by some aspects of diet such as high sodium or phosphate intake.
Mechanisms of increasing arterial stiffness associated with CKD
Increasing arterial stiffness with age
The biomechanical properties of the arteries are largely dependent on the relative quantities of collagen and elastin, the main scaffolding proteins of the extracellular matrix (ECM). Numerous studies have shown that the large arteries stiffen with age with overproduction of abnormal collagen fibres and relative loss of elastin from the ECM.34 The elastin lamellae become sparser with signs of fragmentation and calcification, while collagen molecules progressively acquire cross-links. A major unresolved question is whether these changes are truly time-dependent or reflect exposure to the risk factors listed below. Similarly, the possible effect of the age-related decline in glomerular filtration rates on arterial stiffness remains unknown. Interestingly, recent experimental evidence suggests that nitric oxide-donating drugs may reverse the relative loss of elastin that occurs with age35 and improve endothelial function and cardiac hypertrophy independent of blood pressure.36 These agents may thus have some potential to prevent or reverse the increasing arterial stiffness associated with ageing and CKD.
Alterations in the extracellular matrix
Evidence of altered ECM structure in CKD comes from studies of subtotally nephrectomised rats in which aortic wall thickness was significantly greater than in controls.37 ECM volume was increased, elastic fibres were smaller and collagen ‘islands’ were evident. VSMC were larger and greater in number with ultrastructural changes suggesting increased secretory activity. No studies have yet examined changes in arterial microstructure in human CKD, but the coronary arteries of patients with CKD show increased medial thickness and lower luminal area than those of non-CKD controls.11
The mechanisms underlying ECM changes in CKD are currently undetermined but matrix metalloproteinases (MMPs) have been implicated. These endopeptidase enzymes regulate the ECM and are produced by vascular and inflammatory cells. Increased MMP production enhances collagen and elastin turnover through enzymatic cross-link degradation,38 causing unravelling and weakening of the ECM. Doxycycline, a non-specific inhibitor of MMP, improved elastic fibre integrity and reduced arterial stiffness in animal models of Marfan syndrome.39 There are data supporting the MMP mechanism in hypertensive patients40 and there is also accumulating evidence in CKD. Vascular MMP activity in patients with ESKD is increased compared with healthy controls,41 highlighting a potential target for future therapeutic intervention with MMP inhibitors.
The role of advanced glycation end products
Irreversible covalent cross-linking of collagen and elastin with carbohydrates or carbonyl compounds through non-enzymatic glycation results in the formation of advanced glycation end products (AGE).42 Such post-synthetic glycation occurs in abundance in impaired glucose tolerance and diabetes mellitus and to a lesser extent with ageing, although it is unclear whether or not this is inevitable or a consequence of exposure to factors such as oxidative stress and inflammation.43 Affected collagen is stiffer and less susceptible to slow hydrolytic degradation; glycation may also influence arterial stiffening through generation of reactive oxidant species and nitric oxide deactivation, promoting endothelial dysfunction.44
Levels of circulating AGE correlate directly with serum creatinine in patients with diabetic and non-diabetic CKD.45 46 Advanced glycation end products accumulate in ESKD as demonstrated by skin autofluorescence and are independently associated with increased aPWV47 and with mortality. Although there is some suggestion that AGE are implicated in the development of arterial stiffness in CKD, this finding is not universal.46 Hypertensive subjects and older patients treated with AGE cross-link breakers demonstrate significant reductions in arterial stiffness and endothelial dysfunction.48 49 Further studies are required to determine whether dietary AGE restriction or the use of AGE cross-link breakers might be an effective method of reducing arterial stiffness in CKD.
Endothelial dysfunction
Endothelial dysfunction, characterised by impaired endothelium-dependent vasodilator activity or enhanced endothelium-dependent vasoconstriction, is strongly associated with increased arterial stiffness in healthy individuals.50 Endothelial dysfunction has been demonstrated in patients with CKD compared with controls.51 Although this may reflect relatively high levels of oxidative stress and of risk factors such as hypertension, reduced renal clearance of uraemic toxins, such as asymmetrical dimethylarginine (ADMA), may also contribute.52 ADMA, together with its structural isomer symmetrical dimethylarginine, inhibits nitric oxide synthesis dose-dependently in vitro and increases basal vascular tone and blood pressure in humans.52 53 High plasma ADMA concentrations are associated with, and predict progression of, increased carotid intima-media thickness in ESKD.54
Endothelin peptides are synthesised by endothelial cells and are powerful vasoconstrictors acting on VSMC. They are implicated in the pathogenesis of several cardiovascular conditions and the progression of CKD.55 Infusion of endothelin-1 in healthy humans to increase plasma levels to those seen in ESKD is associated with significant increases in aPWV, central systolic pressure and pulse pressure.56 Short-term endothelin-A receptor antagonism in non-diabetic CKD reduces proteinuria and arterial stiffness independently of blood pressure lowering.57 Long-term effects of endothelin antagonism on arterial stiffness and cardiovascular risk in CKD are yet to be determined, but there is emerging evidence that endothelin antagonists may be of value in treating resistant hypertension, perhaps through direct effects on arterial stiffness.58
Replenishment of glutathione using N-acetylcysteine, a cheap and well-tolerated food supplement, has been shown to improve endothelial function and cardiovascular outcomes in two small studies of patients with ESKD,59 60 and further assessment of its potential in reducing arterial stiffness is warranted.
Although it is accepted that endothelial dysfunction promotes arterial stiffening, a study of cultured endothelial cells in vitro suggests that stiff arteries themselves further reduce nitric oxide bioavailability through diminished expression of endothelial nitric oxide synthase61; arterial stiffness may therefore be self-perpetuating. Novel agents such as ghrelin appear to improve endothelial dysfunction62 and offer potentially promising avenues for further investigation in CKD.
Chronic inflammation
Although commonly regarded as a risk factor for atheroma, a clear association between inflammation and arterial stiffness exists, as demonstrated by studies of conditions characterised by chronic systemic inflammation including rheumatoid arthritis and CKD63 64 as well as studies of inflammatory markers and aPWV in the healthy population.65 More specifically, aortic inflammation, as assessed using positron emission tomography imaging, has recently been shown to influence aPWV.66 The long-term use of immunosuppressive agents in inflammatory disorders is associated with a reduction in surrogate markers of cardiovascular risk67; this may be a potential avenue for treatment, although caution is required in view of the associated complications of such treatment.
Inflammatory degradation of ECM elastin has recently been shown to accelerate arterial calcification in animal CKD models,68 prompting potential exploration of the role of immunosuppression and the selective inhibition of elastase enzymes as therapeutic interventions in arterial stiffness reduction.
The renin–angiotensin–aldosterone system (RAAS)
Angiotensin II is a powerful vasoconstrictor but also promotes inflammation by stimulating VSMC to generate intracellular superoxides and inflammatory cytokines69; furthermore, it induces vascular remodelling through VSMC hypertrophy and proliferation, increased collagen synthesis and increased production of MMP.70 This ECM remodelling can be controlled by angiotensin converting enzyme inhibitors (ACEI).71 ACEI and angiotensin II receptor blockers (ARB) also inhibit AGE formation in vitro dose-dependently, possibly by reducing generation of reactive oxygen species and reactive carbonyl compounds.72
Short- and long-term inhibition of the RAAS with ACEI and ARB treatment is associated with reductions in arterial stiffness but is almost invariably accompanied by blood pressure reduction.71 73 The relative importance of blood pressure lowering is difficult to distinguish from the direct tissue effects outlined above. In a longitudinal study of patients undergoing haemodialysis treated with ACEI, blood pressure lowering combined with decreased aPWV was associated with reduced all-cause and cardiovascular mortality.74 Such mortality benefits were absent in subjects with unaltered aPWV despite blood pressure reduction, lending some support to the theory that the RAAS inhibition reduces risk through a blood pressure-independent mechanism in ESKD. In addition, a pilot study of 25 stage 2 and 3 patients with CKD demonstrated that ARB treatment improved small-artery compliance.75
Aldosterone levels, which frequently remain elevated despite treatment with ACEI and ARB, are correlated with arterial stiffness in hypertensive men independently of blood pressure.76 Aldosterone increases arterial stiffness independently of wall stress in subtotally nephrectomised rats given high-salt diets77 and these effects are inhibited by the mineralocorticoid receptor (MR) antagonist eplerenone. MR activation is associated with endothelial dysfunction and activation of VSMC genes involved in vascular fibrosis, inflammation and calcification.78 Additionally, aldosterone upregulates expression and sensitivity of vascular angiotensin receptors in rats.79 The MR antagonist spironolactone inhibits angiotensin II-mediated VSMC proliferation in vitro, reduces aortic and myocardial collagen accumulation80 and reduces oxidative stress and endothelial dysfunction.81 In subtotally nephrectomised rats, spironolactone reduces proteinuria, arterial pressure and cardiac hypertrophy.82 These studies highlight the importance of aldosterone in the development of cardiovascular and renal injury in animal models of CKD.
There are limited data demonstrating the influence of MR antagonism on arterial stiffness in human CKD. In a randomised placebo-controlled trial, the addition of spironolactone to ACEI/ARB treatment in patients with stage 2 and 3 CKD significantly reduced arterial stiffness and left ventricular mass, supporting the hypothesis that aldosterone is a major mediator of arterial stiffness and LVH in CKD.83 There was, however, a significant reduction in blood pressure such that a blood pressure-lowering effect on arterial stiffness could not be excluded. Larger studies are required to determine whether such treatment is associated with improved clinical outcomes.
A recent meta-analysis of RAAS blockade with either ACEI or ARB in CKD revealed a significant reduction in cardiovascular outcomes and incidence of heart failure in comparison with placebo, although no reduction in cardiovascular or all-cause mortality was noted.84 The mechanism by which this improvement in cardiovascular outcome arises is undetermined, but an assessment of arterial stiffness parameters in such patients may prove to be highly informative.
Diet
Observational studies highlight the importance of environmental factors such as high-salt diets in the development of hypertension and arterial stiffness.85 Dietary sodium enhances age-related vascular changes by promoting VSMC hypertrophy and increased VSMC tone; it also increases collagen cross-linking and facilitates aldosterone-induced oxidative stress and inflammation.85 In the presence of aldosterone, small increases in plasma sodium concentration decrease nitric oxide release and increase endothelial cell stiffness in vitro.86 Restricting dietary sodium in hypertensive subjects effectively reduces arterial stiffness.87 The Western diet is relatively rich in dietary oxidants, AGE and bioavailable phosphate.88 These substances undergo renal metabolism or excretion and therefore accumulate in CKD. The influence of dietary factors such as sodium, phosphate and pro-oxidant compounds on arterial structure and function in subjects with and without CKD is remarkably poorly understood and is a fertile area for future research.
Vascular calcification and disorders of bone and mineral metabolism
Vascular medial calcification is prominent in CKD and has an important role in the pathogenesis of arterial stiffness. The extent of arterial calcification correlates with severity of arterial stiffness independently of age and blood pressure in ESKD and CKD89 and is a strong predictor of all cause and cardiovascular mortality in ESKD.90
Vascular calcification refers to deposition of calcium phosphate mineral (hydroxyapatite) in cardiovascular tissues. Arterial calcification has been long thought to be a passive process, but there is now compelling evidence that it is actively regulated, involving direct osteogenic gene activation together with suppression of calcification inhibitors. Exposure of VSMC to high concentrations of intracellular calcium and phosphate in vitro results in their phenotypic switch to an osteogenic cell type with upregulation of genes promoting matrix mineralisation and calcium deposition.91 Osteogenic differentiation is driven by upregulation of transcription factors such as core-binding factor α1 (cbfa1) and bone morphogenetic protein, which control expression of osteogenic proteins such as osteocalcin, osteonectin and alkaline phosphatase.
Upregulation of transcription factors is induced by hyperphosphataemia, which is prevalent in advanced CKD. A sodium-dependent phosphate co-transporter, Pit-1, facilitates movement of inorganic phosphate into VSMC, which stimulates cbfa1 expression dose-dependently.91 Osteogenic proteins are also expressed in VSMC following their exposure to uraemic serum in vitro92; this occurs independently of phosphate concentration, suggesting that the uraemic milieu, perhaps through oxidative stress, directly induces vascular calcification.
Loss of inhibitors of mineralisation such as fetuin A, osteoprotegerin and matrix G1a protein is associated with progressive arterial and ectopic soft tissue calcification. Levels of matrix G1a protein are inversely correlated with severity of coronary artery calcification.93 In a cross-sectional study of patients with ESKD, fetuin A levels were lower than in healthy controls and were associated with increased cardiovascular and all-cause mortality.94 The loss of these inhibitors in vivo may allow calcification to occur at relatively low phosphate concentrations, as suggested by the association of modestly elevated serum phosphate (still within the reference range) with greater prevalence of coronary, aortic and valvular calcification independent of serum vitamin D and parathyroid hormone levels in studies of patients with CKD.95
Calcium-based phosphate binders and vitamin D supplementation used in the treatment of bone and mineral disorders associated with CKD may contribute to hypercalcaemia and subsequent soft tissue calcification. Vitamin D treatment, however, is associated with reduced cardiovascular mortality in observational studies of ESKD.96 This may be partly explained by reduced vascular calcification through suppressed cbfa1 synthesis.97 Hyperparathyroidism, commonly present in advanced CKD, may also contribute to vascular calcification. Parathyroid hormone receptors are present on VSMC, and parathyroidectomy is associated with reduced calcium deposition.98 Hyperparathyroidism is strongly associated with hypertension, increased arterial stiffness, LVH, cardiac fibrosis, impaired cardiac contractility, impaired endothelial function and cardiovascular mortality.98
Serum phosphate levels still within the reference range are associated with cardiovascular mortality in the general population and in patients with CKD, a renal transplant and ESKD.88 99 In animal CKD models, high dietary phosphate and hyperphosphataemia induce cardiac fibrosis and arterial wall thickening.100 The importance of vascular calcification as a determinant of arterial stiffness suggests that treatments that suppress or inhibit this process may be highly effective in maintaining arterial function. There are a variety of therapeutic targets but powerful phosphate binders are already available. The results of studies determining their influence on markers of vascular calcification and parameters of arterial stiffness may have profound implications for the management of cardiovascular risk in the CKD population.
Conclusion
Increased arterial stiffness is associated with structural and functional cardiac abnormalities as well as increased cardiovascular mortality in patients with CKD. CKD is characterised by accelerated ageing of the vasculature, in which age-related changes of increased arterial stiffening are further compounded by a number of uraemic-related processes. By reducing exposure of the vasculature to processes that induce structural and functional change, we may be able to slow the progression of arterial stiffness and decrease the high cardiovascular risk associated with CKD.
References
Footnotes
Funding Genzyme Corporation, 4620 Kingsgate, Cascade Way, Oxford Business Park South, Oxford OX4 2SU, UK.
Competing interests CJF has received lecture and advisory board fees from Genzyme. All authors are recipients of an unrestricted educational grant from Genzyme Corporation. No medical writers were involved in the preparation of this manuscript.
Provenance and peer review Not commissioned; externally peer reviewed.