Article Text

Abstract
Air pollution is a significant cause of cardiovascular morbidity and mortality worldwide. Although the epidemiologic association between air pollution exposures and exacerbation of cardiovascular disease (CVD) is well established, the mechanisms by which these exposures promote CVD are incompletely understood. This review provides an overview of the components of air pollution, an overview of the cardiovascular effects of air pollution exposure, and a review of the basic mechanisms that are activated by exposure to promote CVD.
Statistics from Altmetric.com
Introduction
A wide variety of epidemiological studies conducted over the last decade have demonstrated a strong association between exposure to air pollution and cardiovascular disease (CVD) (reviewed in Brook et al1). Acute exposure has been associated with an increased incidence of myocardial infarction, arrhythmias, heart failure and all cause mortality. Chronic exposure also promotes CVD and all cause mortality to a significantly greater degree than acute exposure. Although the predominant cardiovascular complication of exposure to air pollution is ischaemic heart disease, statistically significant associations are also seen for arrhythmias, heart failure and cardiac arrest. While the association between air pollution and CVD is now commonly accepted, based on a large number of studies, the exact causal agents within the umbrella of airborne pollutants and the pathophysiological mechanisms by which they cause CVD are incompletely understood. Many earlier studies linking air pollution and CVD have been comprehensively reviewed.1 Accordingly, this review will summarise briefly the components of air pollution that have been linked to CVD and will focus on more recent mechanistic studies outlining how ambient particulate air pollution promotes adverse cardiovascular effects in both animal and controlled human exposure models.
There are six major air pollutants currently regulated by the US Environmental Protection Agency (table 1). The gaseous pollutants include carbon monoxide, nitrogen oxides, sulfur dioxide and ozone, while the ambient particulate matter (PM) components are generally subdivided according to size. These subdivisions reflect the distinct chemical and physical properties of different sized particles, most notably their ability to travel into different parts of the respiratory system. Thoracic particles (PM10), consisting of particles smaller than 10 µm, generally do not penetrate beyond the extrathoracic and upper bronchial regions. Fine particles (PM2.5), consisting of particles smaller than 2.5 µm, penetrate into the small airways and alveoli. Ultrafine particles (PM0.1), consisting of particles smaller than 0.1 µm, penetrate into the alveoli and possibly the systemic circulation. Coarse particles, in general, arise from natural sources, such as soil, agriculture, plants, volcanos and surface mining. Fine particles generally arise from combustion, smog, diesel and gasoline sources. Ultrafine particles exclusively arise from diesel and gasoline combustion, and are a direct consequence of man-made activity. Diesel exhaust is a prime contributor to air pollution, and consists of both particulate and gaseous components. The particles are composed primarily of elemental carbon, adsorbed organic compounds, and small amounts of sulfate, nitrate, metals and other trace elements. Diesel particulates can account for up to approximately 90% of PM2.5 in major cities.1
US Environmental Protection Agency regulated air pollution components and cardiovascular effects in humans
While each of the six regulated components of air pollution has been linked to CVDs such as hypertension, myocardial infarction, stroke, heart failure and cardiac arrest (table 1), the evidence for an association between PM exposure and CVD far exceeds that for other components. Ambient particulate air pollution is the ninth leading cause of disability adjusted life-years worldwide, the fourth leading cause in East Asia, and was responsible for 3.2 million deaths worldwide in 2010.2 Interestingly, the effect estimates of air pollution on cardiovascular complications such as myocardial infarction are higher than the effects on lung disorders such as lung cancer and even on all cause mortality, resulting in higher population attributable risk.
Although a significant proportion of ambient PM2.5 is derived from diesel engines, the chemical composition of PM2.5 can vary greatly depending on proximity to other sources such as power generation, industry, agriculture or aviation. To study the mechanisms by which airborne particulates promote CVD, many animal and human studies use either diesel engine exhaust particulates (DEPs) or ambient particulate material concentrated directly from the atmosphere (concentrated ambient particulates (CAPs)). These particulates are introduced into a controlled exposure chamber at a defined dose for a defined length of time. CAPs consist of PM2.5 from traffic related sources such as diesel exhaust, but also from aviation, agricultural and other intermittent industrial sources of PM2.5 that may be located nearby. The advantage of using CAPs is that the PM2.5 exposures closely match the human exposures in the regional population. The main disadvantage is that these exposures are difficult to compare when obtained from different regions because PM2.5 composition will vary widely both regionally and temporally. In contrast, DEPs are well defined but relatively simple in composition. The inflammatory and toxic potential of these particles is influenced by their content of specific environmental components such as endotoxin and metals.
Both acute and chronic exposure to PM can lead to a variety of specific cardiovascular effects. Associations have been reported with exacerbation of ischaemic heart disease,3 heart failure,4 cerebrovascular disease,5 deep venous thrombosis,6 hypertension7 ,8 and cardiac arrhythmias,9 with varying degrees of evidence supporting these associations. Analysis of the specific molecular and cellular mechanisms involved reveals diverse and overlapping effects in which systemic inflammation, oxidative stress, neurohumoral and epigenetic modifications contribute in a variety of ways. The specific mechanisms by which PM promotes CVD are described below.
Mechanisms by which PM exposure promotes CVD
At present, there are three distinct hypotheses to explain the association between PM exposure and CVD, with varying degrees of evidence and consensus.1 The first hypothesis is best supported and asserts that PM entering the lungs provokes an inflammatory response that promotes oxidative stress and is sufficient to promote systemic oxidative stress and inflammation. This pro-inflammatory state is then thought to promote a variety of pathological processes related to CVD, such as increased thrombosis, hypercoagulability, endothelial dysfunction, atherosclerosis progression, insulin resistance and dyslipidaemia.
The second hypothesis is somewhat supported and asserts that pulmonary exposure leads to activation of lung autonomic nervous system (ANS) arcs mediated by transient receptor potential (TRP) channels that then cause ANS imbalance, leading to pathological alterations in vasoconstriction, endothelial dysfunction, hypertension, platelet aggregation, tachycardia, increased heart rate variability and increased arrhythmia potential. The studies implicating TRP channels are largely done with inhibitors but await confirmation with knockout mice.
The third hypothesis is supported by only a few studies and asserts that airborne particulates and/or their constituents inhaled through the lungs directly enter the circulation where they may directly interact with tissue components to promote vasoconstriction, endothelial dysfunction, atherosclerosis, hypertension, platelet aggregation, systemic oxidative stress and inflammation. Evidence has also been accumulating that PM exposure can lead to chemical modification of DNA, resulting in epigenetic dysregulation of gene expression. We will review the evidence in support of these hypotheses from exposure studies in both animal and human subjects (table 2). A working model of how air pollution promotes CVD is presented in figure 1.
Basic pathophysiological mechanisms activated by exposure to particulate matter in animal and human subjects
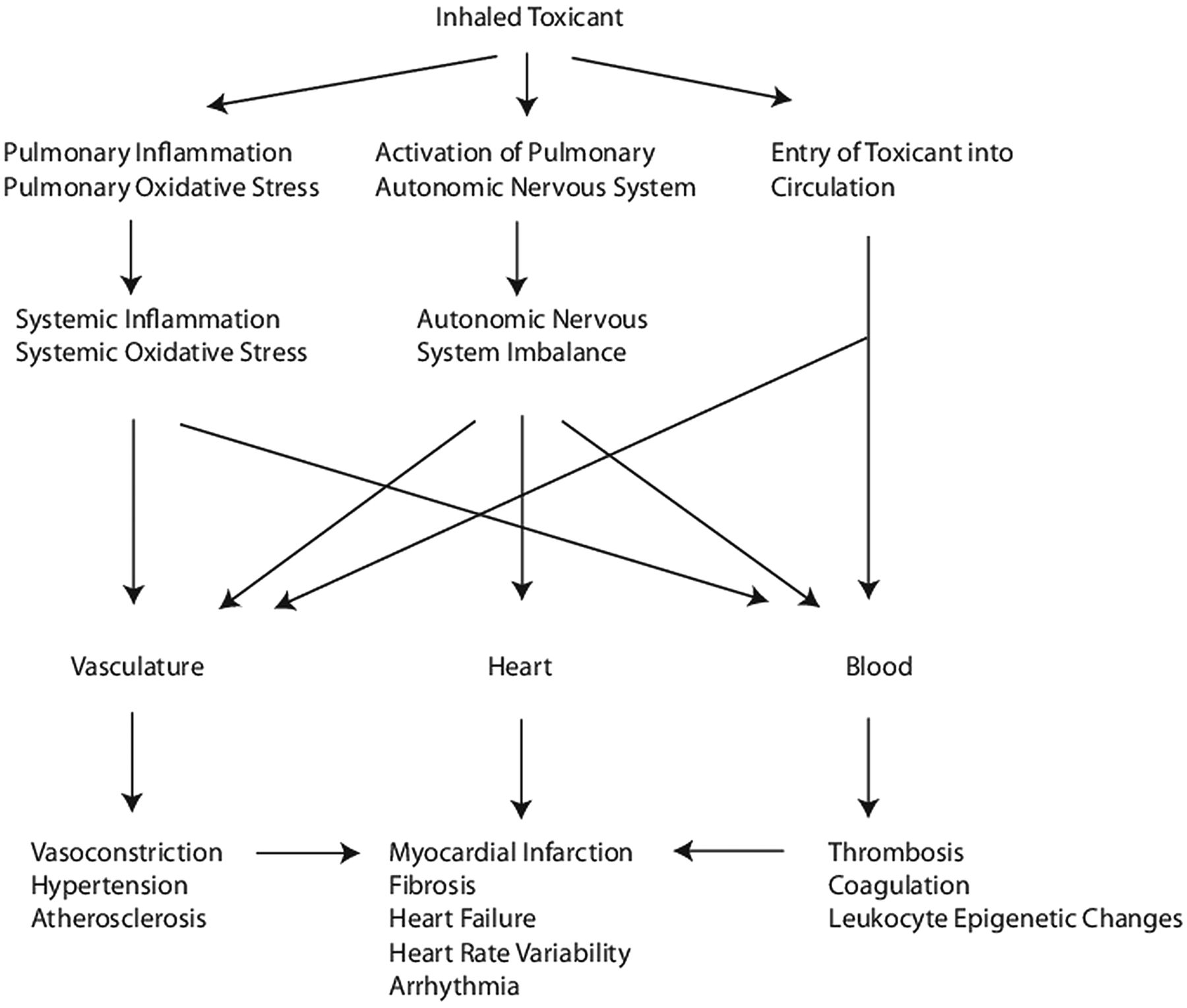
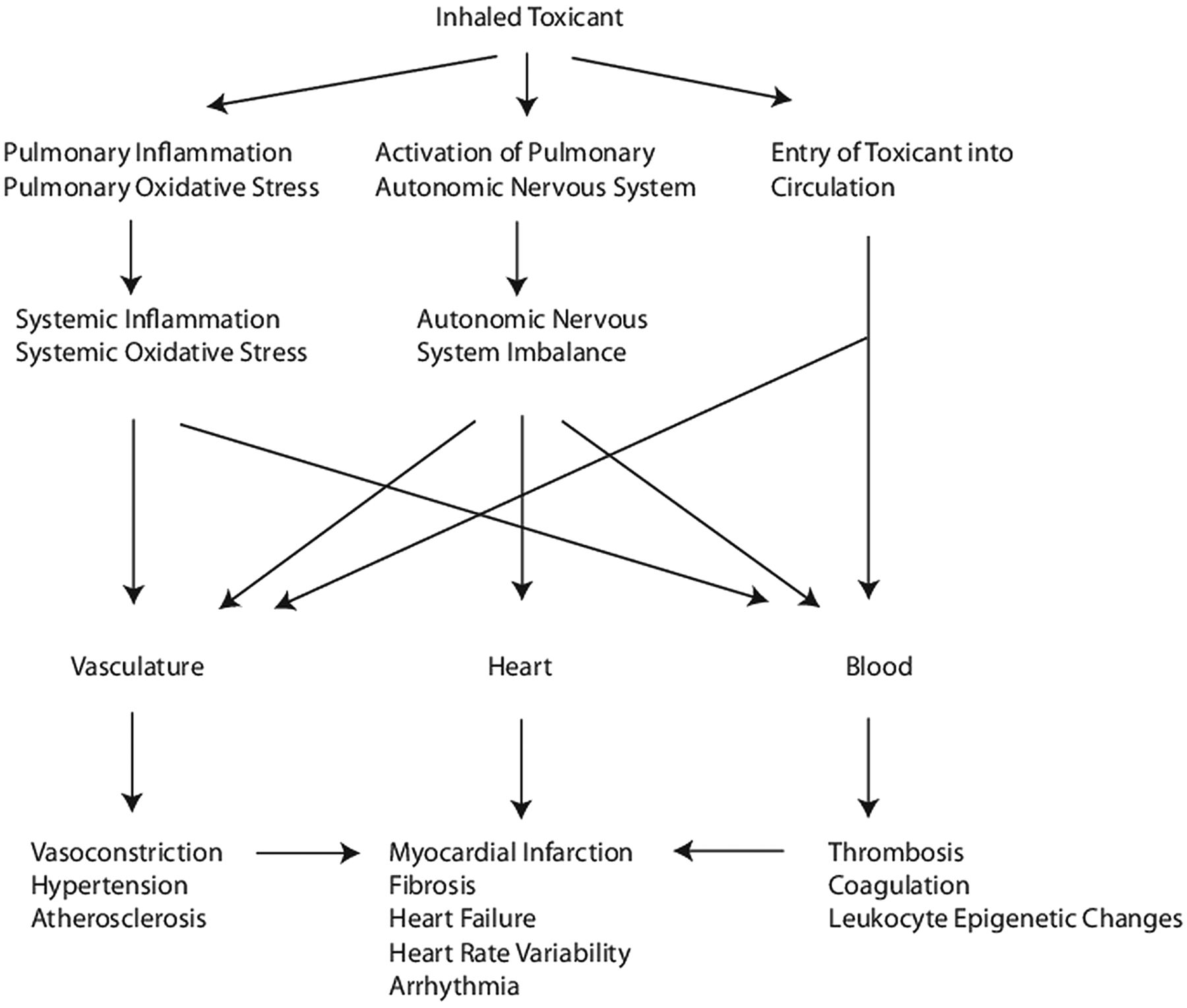{kind=link}
A working model of how air pollution exposure promotes adverse cardiovascular effects.
Systemic inflammation
Diesel exhaust particles have been shown to induce pulmonary inflammation, oxidative stress and pulmonary cell toxicity in both animal models and human subjects, releasing cytokines and other pro-inflammatory mediators into the circulation. In obese mice, CAPs exposure also promotes adipose tissue inflammation, which is thought to propagate systemic inflammation even further (reviewed in Brook et al1). PM2.5 exposure also triggers the appearance of oxidised phospholipids in the lungs that then mediates a systemic inflammatory response mediated through toll-like receptor 4/nicotinamide adenine dinucleotide phosphate (TLR4/NADPH) oxidase-dependent mechanisms.10 Short term exposure studies in humans have also shown evidence of systemic inflammation as measured by concentrations of circulating cytokines such as interleukin-1β, interleukin-6 (IL-6), and tumour necrosis factor α.11 Additional human studies have demonstrated changes in gene expression in peripheral blood mononuclear cells associated with inflammation, oxidative stress and coagulation,12 ,13 suggesting that these mechanisms are clinically relevant. Other human studies have not shown changes in circulating inflammatory mediators, however, indicating that these studies are not always consistent.14
Systemic oxidative stress
Systemic oxidative stress can be induced by circulating cytokines and inflammatory cells that are activated by events induced in the lungs after exposure to PM, or by leaching of soluble components such as oxidised phospholipids,10 thereby promoting oxidative stress in the vasculature and other organs such as adipose tissue, the liver and the heart (reviewed in Brook et al1). Exposure of pregnant mice to DEPs has been associated with placental vascular oxidative stress, implying that systemic oxidative stress is induced.15 Induction of systemic oxidative stress in human subjects exposed to PM under well-characterised conditions has been measured by urinary 8-hydroxy-2′-deoxyguanosine (8-OHdG), a marker of oxidative DNA damage. Urinary 8-OHdG concentrations correlate with reduction in heart rate variability and PM2.5 exposure, confirming the clinical plausibility of this biological mechanism.16 Generation of reactive oxygen species is a hallmark of activated leucocytes and underscores the close link between systemic oxidative stress and systemic inflammation.
Thrombosis and coagulation
In the cardiovascular system, animal studies have suggested that airborne particles can promote hypercoagulability, from either direct translocation of particles into the blood or release of circulating factors, although early studies were done with either intratracheal administration of particulates or with PM10 (reviewed in Brook et al1). A more recent study showed that exposure of mice to inhaled CAPs leads to activation of the sympathetic nervous system and systemic catecholamine release, which in turn activates β2-adrenergic receptor-dependent release of IL-6 from alveolar macrophages to promote a prothrombotic state.17 Controlled human exposure studies have suggested that brief exposure to DEPs promotes an increase in thrombus formation measured ex vivo in a Badimon chamber as well as an increase in platelet–neutrophil and platelet–monocyte aggregates, implying that DEPs induce platelet activation.18
Hypertension
Effects of PM exposure on blood pressure have been variable, probably reflecting differences in methodology but also reflecting a relatively weak effect.1 Long term exposure to CAPs reportedly promotes vascular oxidative stress, increased sensitivity to angiotensin II, activation of Rho/ROCK (Rho associated protein kinase), activation of the sympathetic nervous system, increased expression of endothelin, systemic inflammation, hypothalamic inflammation and systemic hypertension in mice.1 ,19 A controlled exposure study in human subjects indicated that acute exposure to CAPs in conjunction with ozone leads to transient diastolic hypertension, possibly due to autonomic imbalance.8 A more recent study demonstrated that short term, controlled human exposure to PM2.5 CAPs resulted in an increase in systolic blood pressure that correlated with reduced DNA methylation in Alu repetitive elements in circulating leucocytes.7
Vascular dysfunction and atherosclerosis
PM has been shown to have a multitude of vascular effects such as inhibition of endothelium dependent nitric oxide induced vasodilation, enhanced adrenergic induced vasoconstriction, induction of reactive oxygen species, vascular oxidative stress and vascular inflammation, after both acute and chronic exposures in numerous animal studies (reviewed in Brook et al1). Long term exposure to CAPs has been shown to promote experimental atherosclerosis in two different mouse models through increased CD36 expression in plaque macrophages that facilitates greater uptake and accumulation of an oxidised form of cholesterol in atherosclerotic lesions.20 Chronic CAP exposure also promotes inflammatory monocyte egress from bone marrow, production of reactive oxygen species, and subsequent vascular dysfunction through TLR4 activation of NADPH oxidase in mice.10 Controlled exposure in human subjects has also been shown to promote vascular dysfunction acutely through alteration of responsiveness to vasoactive mediators, to reduce endogenous fibrinolysis, and also to enhance exercise induced myocardial ischaemia in patients with coronary disease.21 Confirmation of effects of particulates on atherosclerosis in human patients through controlled exposures is unlikely to be done, given the decades long incubation period for human atherosclerosis.
Heart rate variability and arrhythmias
Exposure of rodents to PM has been shown to decrease heart rate variability and promote arrhythmias, in both acute and chronic settings. This effect correlates with the presence of iron and nickel within the particulates.22 Controlled exposure studies in humans have confirmed that acute exposure to CAPs can cause a temporary reduction in heart rate variability,23 although a more recent study using diesel exhaust showed no effect.24
Myocardial fibrosis and heart failure
A 3-month exposure of mice to CAPs has been shown to exacerbate angiotensin II-induced cardiac hypertrophy in a Rho kinase dependent manner.25 A 9-month exposure of mice to CAPs resulted in increased ventricular size along with systolic and diastolic dysfunction attributable to myocardial fibrosis.26 Interestingly, exposure of pregnant mice to DEPs conveyed a long lasting susceptibility to myocardial fibrosis and heart failure in pups raised to adulthood,15 suggesting that mediators in the maternal circulation could cross the placenta and promote susceptibility in the developing embryo hearts, through a potential epigenetic mechanism. Although epidemiologic studies have linked air pollution exposure to heart failure,4 controlled human exposure studies have not yet demonstrated an effect of particulates on heart function, to the best of our knowledge.
Epigenetic changes and gene expression
Animals exposed to air from steel plants developed mutations in an expanded simple tandem repeat locus analysed in sperm DNA. The sperm DNA was also found to be hypermethylated when compared to DNA from control mice breathing filtered air, and these changes persisted after the exposure was terminated.27 Controlled human exposure studies showing induction of epigenetic changes in the heart, vasculature or immune cells that alter gene expression and mediate cardiovascular effects have been limited; however, a recent controlled exposure study demonstrated an effect of short term particulate exposure on reduction of DNA methylation in Alu repetitive DNA elements in circulating leucocytes that correlated with reduction in blood pressure.7
Clinical and scientific challenges for the future
The epidemiological link between air pollution exposure and CVD is well established, and the global impact is clear. Proper environmental policy will be required to minimise unnecessary exposures and therefore limit the impact of air pollution on the incidence and prevalence of CVD. Healthcare workers can advise patients about their risks to help minimise individual exposure. Future studies identifying molecular pathways involved such as IL-6 and Rho/ROCK may lead to the development of pharmacological agents that will mitigate the damage in individuals who cannot avoid exposure.
Acknowledgments
I apologise to the many investigators whose work could not be included in this manuscript due to space limitations. I thank Michael Rosenfeld and Terry Kavanagh for critical reading of the manuscript. MTC is supported by NIH grant ES023015.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- Cardiac risk factors and prevention
- Heartbeat