Article Text

Abstract
Introduction Anaemia is common in aneurysmal subarachnoid haemorrhage (aSAH) and is a potential critical modifiable factor affecting secondary injury. Despite physiological evidence and management guidelines that support maintaining a higher haemoglobin level in patients with aSAH, current practice is one of a more restrictive approach to transfusion. The goal of this multicentre pilot trial is to determine the feasibility of successfully conducting a red blood cell (RBC) transfusion trial in adult patients with acute aSAH and anaemia (Hb ≤100 g/L), comparing a liberal transfusion strategy (Hb ≤100 g/L) with a restrictive strategy (Hb ≤80 g/L) on the combined rate of death and severe disability at 12 months.
Methods Design This is a multicentre open-label randomised controlled pilot trial at 5 academic tertiary care centres. Population We are targeting adult aSAH patients within 14 days of their initial bleed and with anaemia (Hb ≤110 g/L). Randomisation Central computer-generated randomisation, stratified by centre, will be undertaken from the host centre. Randomisation into 1 of the 2 treatment arms will occur when the haemoglobin levels of eligible patients fall to ≤100 g/L. Intervention Patients will be randomly assigned to either a liberal (threshold: Hb ≤100 g/L) or a restrictive transfusion strategy (threshold: Hb ≤80 g/L). Outcome Primary: Centre randomisation rate over the study period. Secondary: (1) transfusion threshold adherence; (2) study RBC transfusion protocol adherence; and (3) outcome assessment including vital status at hospital discharge, modified Rankin Score at 6 and 12 months and Functional Independence Measure and EuroQOL Quality of Life Scale scores at 12 months. Outcome measures will be reported in aggregate.
Ethics and dissemination The study protocol has been approved by the host centre (OHSN-REB 20150433-01H). This study will determine the feasibility of conducting the large pragmatic RCT comparing 2 RBC transfusion strategies examining the effect of a liberal strategy on 12-month outcome following aSAH.
Trial registration number NCT02483351; Pre-results.
- NEUROSURGERY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Rigorous trial methodology to evaluate the feasibility of conducting a larger trial to establish optimal red blood cell transfusion thresholds in patients with aneurysmal subarachnoid haemorrhage.
The multicentre pragmatic design strengthens future results and generalisability.
To minimise any potential bias from the necessary open-label design, collection of cointerventions and blinded outcome-assessment are being undertaken.
The pilot trial was not designed to test clinically relevant outcomes.
Introduction
Aneurysmal subarachnoid haemorrhage (aSAH) is a devastating illness caused by the spontaneous rupture of a weakened and enlarged artery in the brain. It affects a young population and is a significant cause of premature death and loss of potential life years, at a similar magnitude of ischaemic stroke.1 It is a common neurological reason for intensive care unit (ICU) admission2 and is associated with a mortality rate of about 35% in North America (range 20–70%).3 Less than one-third afflicted make a full recovery4 and 20% of survivors experience significant morbidity5 having an impact on daily living.
Anaemia (haemoglobin (Hb) <100 g/L) affects more than 50% of aSAH patients and is associated with worse clinical outcomes.4 ,6–11 Preclinical studies in brain injury suggest that red blood cell (RBC) transfusion to treat anaemia optimises oxygen delivery in this specific setting.4 However, RBC transfusions are not without risk and are a limited and expensive resource.12 The limited evidence examining the association between RBC transfusion and clinical outcome from aSAH is derived from few observational studies with conflicting results and significant methodological limitations.5 ,7 ,9 ,10 ,13–17 Only one small trial compared two transfusion targets in aSAH but was underpowered to examine clinically important outcomes.18 Despite this absence of evidence, current aSAH management guidelines include a recommendation to consider RBC transfusion in anaemic patients at risk for cerebral ischaemia, but do not suggest transfusion thresholds to guide clinicians.19 ,20 These recommendations are in contrast with evidence from randomised controlled trials (RCTs) in other critically ill adult and paediatric populations which support a more restrictive RBC transfusion approach.21 ,22
Although the biological rationale and current recommendations for treating aSAH patients support a higher transfusion threshold (liberal strategy), the clinical evidence is lacking to substantiate these recommendations. Current stated and observed practice from surveys23 and our own observational work suggest a more restrictive approach to transfusion (lower haemoglobin); similar to other critical care patients. However, unlike other critically ill patients, brain injury and the sequelae that follow (eg, vasospasm and delayed cerebral ischaemia) may make these patients more susceptible to the decreased oxygen delivery associated with a lower transfusion threshold. Considering this obvious paradox and confliction, there is pressing need to generate high-quality evidence to guide clinical RBC transfusion practices in aSAH. The clinical impact of varied transfusion thresholds in aSAH has never been studied in a large and rigorous randomised trial. In collaboration with the Canadian Critical Care Trials Group (http://www.ccctg.ca), we aim to conduct such an RCT comparing two RBC transfusion strategies in adult patients with aSAH powered for clinically relevant outcomes. To inform and justify our large trial, we are conducting a pilot RCT to assess feasibility and strengthen the design of the large-scale trial.
Methods and analysis
Study design
The Aneurysmal SubArachnoid Hemorrhage—Red Blood Cell Transfusion And Outcome: a pilot randomised controlled trial (SAHaRA Pilot Trial) is a multicentre open-label randomised controlled pilot trial in patients with an acute aSAH at five Canadian academic tertiary care hospitals. To reduce bias from the open-label design, outcome assessors will be blinded to the treatment assignments.
Objectives
Primary objective: To evaluate the feasibility of reaching an optimal randomisation rate of at least one patient per month per centre over the pilot trial period.
Secondary objectives: (1) To evaluate the feasibility of obtaining: (a) at least 90% adherence to the allocated study transfusion thresholds; (b) at least 90% adherence to the study RBC transfusion protocol; and (c) ≥95% clinical outcomes measures (modified Rankin Scale (mRS), Functional Independence Measure (FIM) and EuroQOL Quality of Life Scale (EQ5D)) at 6 and 12 months.
Patient population
To facilitate randomisation into the pilot trial, a subset of patients most likely to meet randomisation criteria will be identified (Screen Eligible Patients).
To be screened eligible for enrolment, patients must meet all inclusion criteria and no exclusion criteria.
Inclusion criteria:
Age ≥18 years at the time of presentation
First ever episode aSAH
Confirmed aSAH diagnosis: as confirmed by treating neurosurgeon or neurointerventionalist and supported by blood in subarachnoid space (demonstrated on cranial imaging or cerebrospinal fluid positive for xanthochromia) that is the result of a ruptured saccular aneurysm (confirmed by cranial imaging—CT, magnetic resonance or catheter angiogram)
Incident Hb ≤110 g/L within 14 days following aSAH (defined by first day of hospital presentation)
Exclusion criteria
Physician and or next of kin decision to withdraw/withhold critical care at the time of enrolment
Active bleeding with haemodynamic instability at the time of enrolment
Patients with contraindication or known objection to blood transfusions
SAH due to causes other than saccular aneurysm rupture including mycotic, traumatic and dissecting aneurysms, and aneurysms associated with arteriovenous malformations
Our exclusion criteria are in place to prevent enrolling patients who: (1) would not benefit from the intervention; (2) object to the intervention; and/or (3) have sustained a bleed due to mechanistically different causes whose pathophysiological properties are not necessarily shared with aSAH.
Screen eligible patients who experience an incident Hb ≤100 g/L within 14 days following aSAH will be randomised.
Randomisation and allocation concealment
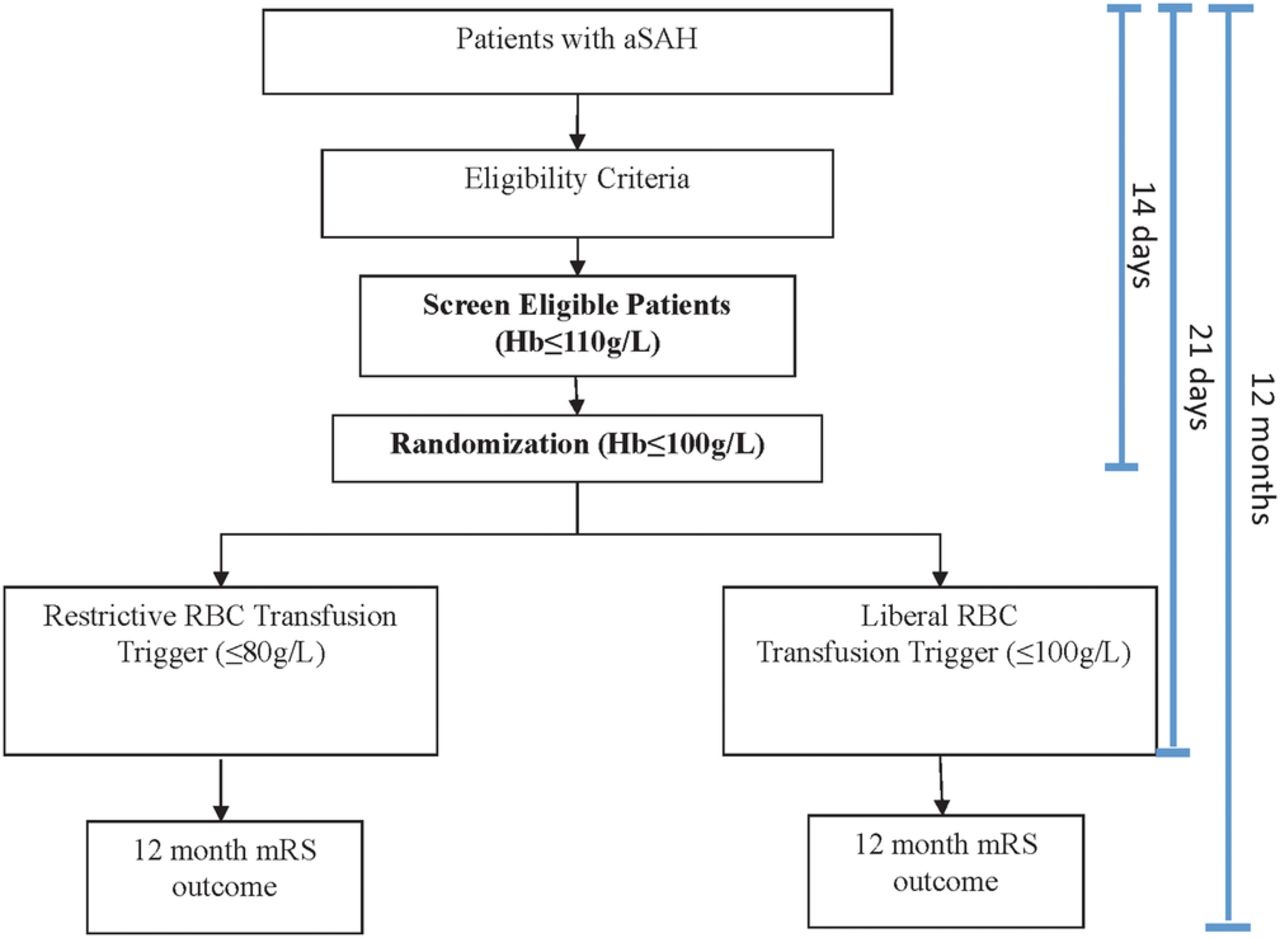
Figure 1 provides a schematic description of the trial design. Local research coordinators will screen each patient admitted to either the ICU, intermediate care unit or step-down unit (where applicable) or neurosurgical inpatient unit in the setting of aSAH for up to 14 days after the qualifying bleed. Enrolment and randomisation over 14 days is necessary as previous work has demonstrated that the negative effect on outcome was most pronounced in patients with anaemia between days 6 and 11.24 Further, our observational study demonstrated that 95% of incident anaemia occurred within the first 14 days, and that 97.4% did so while admitted to a high-acuity unit.25 The risk of new-onset vasospasm, a significant threat to morbidity and mortality in this population is highly unlikely to begin after 14 days, but its duration may surpass this period.19 A Screen Eligible period is essential to focus study resources on the group of patients most likely to be randomised, to capture the first occurrence of anaemia (to minimise any exposure time below their allocated transfusion threshold) and to optimise the randomisation rate. The study team will screen daily haemoglobin values (or more frequent as clinically indicated and/or as deemed by treating team) of Screen Eligible Patients.

SAHaRA trial design.
Patients meeting eligibility criteria (or their substitute decision maker) will be approached for consent by the site research coordinator in accordance with standard local procedures as approved by each local REB and in accordance with Good Clinical Practice. A mixed consent model (a priori and deferred consent models), pending on local REB approval, will be used. A web-based randomisation system maintained at the Coordinating Center will be used to allocate treatment assignments. Under the guidance of the site principal investigator or research coordinator, the participant's eligibility criteria will again be confirmed with a checklist using a web interface. On meeting the randomisation criteria, patients will be randomised in a 1:1 manner to either liberal (intervention) or restrictive (control) RBC transfusion strategy groups. A schedule of the random treatment allocations, stratified by centre will be prepared by an independent biostatistician at the Coordinating Center. All investigative team members will remain blinded to the allocation schedules.
Intervention
Patients fulfilling the eligibility criteria will be randomised to either a liberal or restrictive RBC transfusion strategy.
Intervention group: liberal RBC transfusion strategy
In this intervention group, an RBC transfusion will be triggered by a haemoglobin level of ≤100 g/L over the first 21 days in hospital following aSAH.
Control group: restrictive RBC transfusion strategy
For patients randomised to this group, an RBC transfusion is permitted once a haemoglobin level of ≤80 g/L is observed over the first 21 days in hospital following aSAH. RBC transfusion will not be mandatory under this threshold, ‘usual care’ rather will prevail and the decision to and timing of transfusion will be left to the discretion of the treating team.
Both groups
All RBC transfusions will be a single unit unless the patient has an active blood loss associated with haemodynamic instability. In stable non-bleeding patients, a second unit of RBCs should only be given if a measured post-transfusion haemoglobin level remains below the patient's assigned threshold.
Justification of the two triggers
Intervention: Liberal RBC transfusion trigger (100 g/L): supported by:
Physiological evidence that RBC transfusion increases oxygen delivery and cerebral tissue oxygen tension.26–28
Among SAH patients with haemoglobin <110 g/L, compared with induced hypertension and fluid bolus, RBC transfusion was the only intervention demonstrated to significantly reduce (47%) the number of cerebral regions with low oxygen delivery per patient. Among those with low global oxygen delivery, RBC transfusion resulted in a significant larger rise in global oxygen delivery.26
A small physiological study of aSAH patients (N=8) demonstrated stable cerebral blood flow, an increase in oxygen delivery and a decrease in the oxygen extraction fraction with an RBC transfusion at a haemoglobin level of <100 g/L.29
A haemoglobin level of <100 g/L was associated with brain tissue hypoxia and metabolic distress compared with those with haemoglobin >100 g/L.30
The maximum threshold haemoglobin to trigger RBC transfusion in the context of a study among the 531 intensivists, neurointensivists and neurosurgeons surveyed was 100 g/L.23
Control: Restrictive RBC transfusion trigger (80 g/L): supported by:
In a survey of 531 practicing intensivists, neurointensivists and neurosurgeons in North America, the median haemoglobin to trigger a transfusion ranged from 75 to 80 g/L depending on SAH grade.23
Among practicing intensivists, neurointensivists and neurosurgeons, the lowest acceptable threshold haemoglobin to trigger an RBC transfusion was 70 g/L in >70% of respondents.23
A Canadian multicentre observational study (N=434) conducted in four academic centres in 2012 and 2013 completed by the SAHaRA study team demonstrated that the median pretransfusion haemoglobin was 79 g/L (IQR 74–93 g/L).31
A transfusion trigger of 100 g/L has previously been shown to be safe in an aSAH population.18 The allocated transfusion strategy will be applied from the time of randomisation to day 21 after the original bleed, death or hospital discharge, whichever comes first. The first 21 days following aSAH represent the period of greatest vulnerability to the direct consequences of aSAH, and the sequelae, including vasospasm, that follow.
Outcomes
Primary outcome
The primary feasibility end point is the number of patients randomised per centre per month over the study period. We expect, among patients suffering from aSAH and anaemia, 1.5 patients/month at each of five sites to be randomised into the trial. We reason that an optimal randomisation rate of 1.5 participants/month/site and as low as one participant/month/site will be necessary to demonstrate the feasibility of conducting the larger planned trial (figure 2). This outcome is objective, readily measurable and feasible based on data generated from a cohort study conducted by the authors.

{kind=link}
{kind=link}
Effect of recruitment rate on study duration: projects 5 sites with staggered initiation of enrolment.
Secondary outcomes
Transfusion threshold adherence will be described as the proportion of ‘per protocol’ RBC transfusion events. A transfusion threshold event is defined as an occurrence which starts when a haemoglobin value is measured at or below the allocated threshold for the first time since the previous event and ends when one of the following occurs: (1) an RBC transfusion is administered; or (2) a repeat haemoglobin is obtained above the allocated threshold within 24 hours of the original measure.
Transfusion threshold non-adherence will be considered to have occurred with any of the following: (1) an RBC transfusion occurs before a transfusion threshold is crossed; or (2) in the liberal arm, a transfusion is not given following a threshold crossing. Transfusion threshold non-adherence will be considered a deviation if: (1) the early transfusion occurs within 5 g/L above the allocated threshold (eg, ≤105 g/L for the liberal arm or ≤85 g/L for the restrictive arm) or, (2) in the liberal arm, an RBC transfusion does not occur for a haemoglobin measure up to 5 g/L below the threshold (ie, a transfusion does not occur for an Hb of 95–100 g/L). All other threshold event non-adherences that are greater or less than 5 g/L for the liberal threshold and >5 g/L below the restrictive threshold will be considered a protocol violation. Transfusion outside of haemoglobin thresholds for symptomatic anaemia or in the event of an active blood loss associated with haemodynamic instability, as defined by the treating team, will be recorded, but not considered a protocol violation. Details on non-adherence (date and haemoglobin level prior to transfusion) and reasons for non-adherence (eg, physician preference, patient instability, active bleeding, safety concern) will be recorded.
B. RBC transfusion protocol adherence: The SAHaRA investigators recognise the importance of minimising exposure time below the allocated transfusion threshold and thus every effort shall be put forth to administer the transfusion expeditiously. For the pilot, we endeavour not to exceed 6 hours from transfusion threshold event to transfusion initiation, in keeping with revascularisation time performance measures in stroke literature.32 ,33 The median time (and IQR) to RBC transfusion will be described. Transfusion protocol adherence will be defined as the proportion of RBC transfusions that are initiated within 6 hours. Non-adherence will be considered to have occurred if there is a delay of more than 6 hours between transfusion threshold event and transfusion initiation. Transfusions occurring between 6 and 24 hours will be considered a protocol deviation and >24 hours from the threshold event will be considered a violation.
C. Clinical outcome ascertainment will include ability to capture vital status at discharge, mRS score at 6 and 12 months and the FIM and the EQ5D at 12 months. The mRS, FIM and EQ5D will be completed by assessors blinded to participant treatment allocation. Each of the outcome assessment measures have been selected because they examine different aspects of the three primary levels of body function and stroke rehabilitation (impairment, activity and participation)34 and are specifically validated and recommended outcome measures in stroke research.35 The mRS is used as the outcome measure over mortality as it includes a spectrum allowing consideration of severe disability and mortality together as both are highly clinically significant. Neurological outcome as assessed by mRS is a common outcome in the aneurysmal SAH literature9 ,18 ,36–39 and is readily interpretable in this community. It takes <15 min to administer, and can be completed using a structured interview40–42 or as a telephone interview.43 The FIM is a validated44 ,45 tool consisting of 18 items that assesses 13 different motor and 5 cognitive tasks previously tested in stroke populations, including aSAH,44 ,46 and has an established minimal clinical important difference in this population.47 It has demonstrated excellent consistency in inter-rater reliability and internal consistency specifically in neurological disorder populations. It is easy to administer and is validated for use by telephone and via proxy respondents.34 The EQ5D is a short and simple two-part questionnaire that may be self-administered, completed by interview or via a proxy respondent, and is used to value and describe health states.34
Baseline characteristics, cointervention, outcome assessment and follow-up
A secure web-based prepiloted data collection form will be maintained by the host centre and utilised for data entry and management. Important baseline characteristics (table 1) will be captured at the time of enrolment for comparison between the two study groups to demonstrate the effectiveness of randomisation. In this trial, patient management outside of RBC transfusion will be left to the discretion of the treating team and in accordance with practice guidelines19 which will be made available to all participating centres and clinicians. All major cointerventions (eg, vasospasm, aneurysm and blood pressure management—table 2) will be carefully documented with daily record by the investigative team. Other clinical outcomes being collected include incidence and severity of vasospasm, incidence of cerebral infarction not directly related to complication from securing aneurysm, need for intubation, tracheostomy, percutaneous gastrostomy tube and/or ventricular shunt and ICU and hospital lengths of stay.
Important baseline characteristics (from time of enrolment and randomisation) to be prospectively collected
Important cointerventions to be prospectively collected
Descriptive metrics will be used to measure feasibility, including the primary outcome of randomisation rate. These data will be gathered prospectively at each study centre by a trained and qualified study nurse or practitioner using an electronic case report form. The number of eligible but not enrolled patients will be tracked, and reasons for non-enrolment will be recorded. Protocol adherence will be assessed prospectively by trained study personnel and all episodes of non-adherence to protocol will be adjudicated by three members of the steering committee, blinded to clinical outcome. Protocol adherence will be reported as a ratio of total correct transfusion threshold events to a combination of total number of transfusion events (needed or not needed per protocol) and total number of missed transfusion non-adherence events. Feasibility of outcome assessment will be measured by the ability to obtain the defined outcome measures at the prespecified time periods. The three outcome measurement instruments (mRS, FIM and EQ5D) will be implemented by a trained and qualified study coordinator blinded to the intervention according to the defined schedule (table 3). Vital status at discharge and adverse events will be captured using a case report form prospectively by the site investigator or the research coordinator.
Schedule of assessments
Executive and Steering Committee roles and responsibilities
The Ottawa Hospital Research Institute is the host and trial method's centre. The eight members of the SAHaRA Executive Committee will oversee all aspects of the study as well as larger research agenda business and will meet quarterly (via teleconference) to discuss any challenges. The Executive Committee will also contribute to the formulation of the analytical plan, data interpretation and the drafting and revisions of future manuscripts. The Steering Committee will consist of all coinvestigators participating in the trial. They are responsible for all aspects of study initiation and conduct at their respective sites. These include timely submissions to research ethics boards and supervision of the research coordinators who will screen, enrol, consent and collect data during the pilot, monitoring of recruitment and monitoring adherence to study protocol, and any operational challenges associated with the pilot RCT.
Ethics, confidentiality and data monitoring body
The study protocol has been approved by the host centre (Ottawa Health Science Network Research Ethics Board (REB)—OHSN-REB 20150433-01H). The intervention and control arm of the trial are part of usual care in many centres, and thus, the research risk to participants is minimal. Safety considerations are addressed within the protocol, and allow for individualised care where needed. In addition to potentially intervention-related adverse event reporting, predefined expected adverse events will be prospectively monitored and include acute respiratory distress syndrome, cardiovascular failure, cardiac ischaemia/infarction, venous thromboembolic events, septic shock, hospital-acquired infections and transfusion reactions.
All participant data will be deidentified to ensure confidentiality and through the assignment of an anonymous identifier by the web-based randomisation tool and data collection form. All data will be collected and stored in firewall-protected, secure servers at the host centre according to institutional and REB policy and in accordance with Good Clinical Practice.
A three-member independent Data Safety Monitoring Committee (DSMC) has been assembled and will oversee the progression of ascertaining the pilot objectives and all trial safety aspects according to a prescribed schedule, DSMC Charter and GCP reporting.
Sample size
A sample size of 60 patients will allow us to evaluate enrolment rate averaging 1.5 patients per month per centre with 5 centres over a 1-year study period. Based on our cohort study, we expect that 90 eligible patients will need to be screened into the study to achieve a randomised sample of 60 patients (ie, more than 2/3 of patients with a haemoglobin of ≤110 g/L had a nadir of 100 g/L or less). All five proposed pilot trial sites are academic tertiary care centres with ∼60–120 aSAH admissions per year. Our sample size will also allow the demonstration of a protocol adherence rate of 90% with a 95% CI of 82.4% to 97.6%.
Analytical plan
Descriptive analyses: Baseline characteristics and management data will be presented with means (continuous measures) or proportions (categorical or ordinal data) with 95% CIs.
Primary outcome: Using descriptive statistics, the median randomisation rate (patients/month) overall and per centre over the study duration will be calculated and reported with IQR. Only actual months where each centre is actively recruiting patients will be considered in the analysis (ie, staggered start up across centres). Figure 2 demonstrates the effect of different randomisation rates on study duration and hence feasibility. A rate of <1 patients/month per centre will prompt a site review of the screening log to examine reasons for missed eligible patients and to discuss how to increase recruitment rate. Achieving our internal pilot primary objective of a randomisation rate of 1.5 patients per month per centre will allow us to complete the large trial in 3.5 years with 10 recruiting centres.
Secondary outcomes: Secondary feasibility outcomes will be reported using descriptive statistics. Protocol adherence will be reported as a proportion as described above. Overall protocol adherence as well as adherence in the two individual study arms will be reported. Given the internal pilot design, with the plan to include these data in the large trial if no substantial changes to the protocol are made after the pilot trial, clinical outcomes will be described in aggregate using descriptive statistics.
Study timeline
We estimate a study duration of 30 months. Study centre identification is complete. Patient enrolment began in mid-October 2015 and will take 12 months per centre to complete or 16 months total assuming a staggered start (to allow for different lead times for site preparation including contracts and REB approval). The last clinical outcome measure is thus expected at 28 months leaving two additional months for data cleaning and analysis for manuscript preparation.
Dissemination
The results of the SAHaRA Pilot RCT will be disseminated to the participating centres. As an internal pilot RCT, should no significant modification of the protocol be necessary, participant data will be included in the planned larger trial powered to clinically important outcomes. The results of the pilot will be incorporated with the larger trial and submitted to peer-reviewed journals for publication and presented at conferences.
Discussion
The TRICC trial,22 the first rigorous trial comparing different RBC transfusion thresholds in a critically ill patient population, remains significant today and continues to guide management of many ICU populations. However, several subpopulations were under-represented (or not at all) in this study, such that the debate of optimal transfusion threshold continues to plague physicians at certain ICU bedsides. The neurocritically ill, specifically aSAH patients, are such a population. The clinical importance of varied transfusion thresholds in aSAH has never been studied in a large and rigorous randomised trial. The only RCT used transfusion thresholds that differ significantly from stated current practice, and was not powered for clinically meaningful outcomes.18 The need for quality evidence to guide transfusion practice in aSAH has been identified by many influential societies, editorials and practice guidelines.15 ,19 ,20 ,51 The uncritical use of variable thresholds does not advance patient outcome or physician practice.
Accomplishing the feasibility objectives of this pilot trial will ensure the successful completion of the future large trial. A pilot trial powered to feasibility outcomes is the essential initial step in the preparation for and eventual successful completion of the more costly larger trial, powered to clinically relevant outcomes.52 Our multicentre design is essential to demonstrate the feasibility of enrolment and randomisation into the study and across centres. A 12-month enrolment period will enable us to determine the feasibility of recruitment at individual centres. Only an open-label design is feasible in an RBC transfusion strategy trial, given the inability to blind bedside clinicians to haemoglobin levels in the safe management of these patients. Similar open-label trial designs have been successfully completed in RBC transfusion trials involving other patient populations.22 ,53–55 Further, prospective randomised open-label blinded end point (PROBE) designs have been used in multiple successful, practice-changing stroke trials.56–58 To minimise potential bias imposed from open-label treatments, our clinical outcome measures will be completed by a blinded assessor who has not been involved in patient management and is unaware of treatment assignment. We will demonstrate the feasibility of collecting the proposed clinical outcomes of the large RCT (neurological functional outcome using mRS at 6 months and 1 year, as well as the FIM and EQ5D at 1 year) by observing the same follow-up schedule.
The results of the SAHaRA internal pilot trial will directly inform the conduct of and guide the successful completion of the larger RCT. The SAHaRA trial will clarify the role of treating anaemia with RBC transfusion in this unique and vulnerable patient population, and whether that impacts on functional outcome and mortality. We hypothesise an improvement in outcome with the treatment of anaemia which, if substantiated, would dramatically change the management of these patients by intensivists, neurologists and neurosurgeons worldwide. A null result would provide the necessary evidence to the bedside clinician that a restrictive transfusion approach is safe and prevent the unnecessary risk imposed by blood product transfusion that regularly occurs.
Acknowledgments
The authors thank Dr Jacques Lacroix from the Canadian Critical Care Trials Group for a critical review of this manuscript. The authors also wish to acknowledge the administrative support Ms Marnie Gordon and Mr Irwin Schweitzer. MC and FL are recipients of a Salary Support Award from the Fonds de Recherche du Québec—Santé (FRQS). AFT is a recipient of a New Investigator Award from the CIHR.
References
Footnotes
Twitter Follow Almunder Algird @Munderal
Contributors SWE, LAM, DF, MC and AFT conceived the project idea. SWE, LAM, DF, MC, AFT, FL, DG, AA, AK, AT, CL, JS, SM, DD, AB and GP all contributed substantially to the design of the trial and drafting of the protocol. SWE created the first draft of this submission and all authors have provided critical review and approve of this final version.
Funding This work is supported by a Transfusion Science research grant awarded by a Canadian Blood Services and Health Canada in partnership with Canadian Institutes of Health Research (CIHR) Institute of Circulatory and Respiratory Health, competition code 201503OTS.
Competing interests None declared.
Ethics approval Ottawa Health Sciences Network Research Ethics Board.
Provenance and peer review Not commissioned; externally peer reviewed.