Article Text

Abstract
Introduction Ductal carcinoma in situ (DCIS) is a non-invasive non-obligate precursor of invasive breast cancer. With guideline concordant care (GCC), DCIS outcomes are at least as favourable as some other early stage cancer types such as prostate cancer, for which active surveillance (AS) is a standard of care option. However, AS has not yet been tested in relation to DCIS. The goal of the COMET (Comparison of Operative versus Monitoring and Endocrine Therapy) trial for low-risk DCIS is to gather evidence to help future patients consider the range of treatment choices for low-risk DCIS, from standard therapies to AS. The trial will determine whether there may be some women who do not substantially benefit from current GCC and who could thus be safely managed with AS. This protocol is version 5 (11 July 2018). Any future protocol amendments will be submitted to Quorum Centralised Institutional Review Board/local institutional review boards for approval via the sponsor of the study (Alliance Foundation Trials).
Methods and analysis COMET is a phase III, randomised controlled clinical trial for patients with low-risk DCIS. The primary outcome is ipsilateral invasive breast cancer rate in women undergoing GCC compared with AS. Secondary objectives will be to compare surgical, oncological and patient-reported outcomes. Patients randomised to the GCC group will undergo surgery as well as radiotherapy when appropriate; those in the AS group will be monitored closely with surgery only on identification of invasive breast cancer. Patients in both the GCC and AS groups will have the option of endocrine therapy. The total planned accrual goal is 1200 patients.
Ethics and dissemination The COMET trial will be subject to biannual formal review at the Alliance Foundation Data Safety Monitoring Board meetings. Interim analyses for futility/safety will be completed annually, with reporting following Consolidated Standards of Reporting Trials (CONSORT) guidelines for non-inferiority trials.
Trial registration number NCT02926911; Pre-results.
- ductal carcinoma in situ
- active surveillance
- breast cancer
- clinical trial
- non-invasive
- surgery
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Comparison of Operative versus Monitoring and Endocrine Therapy (COMET) is a phase III randomised controlled clinical trial.
Ongoing data collected from women who decline randomisation will provide valuable information about the potential for selection bias/enable the study to be made more generalisable.
The comparator arms are very different from each other.
There exists considerable variation between pathologists in the diagnosis of ductal carcinoma in situ.
Introduction
Ductal carcinoma in situ (DCIS): potential risks and burdens
Annually, approximately 65 million women undergo mammographic screening in the USA at a cost of over 13 billion dollars. Almost one in 1300 mammograms (MMGs) will detect ductal carcinoma in situ or DCIS,1 with more than 50 000 women in the USA alone diagnosed with DCIS each year. Almost all diagnoses are made in completely asymptomatic individuals.2 Without treatment, it is estimated that only 20%–30% of DCIS will progress to invasive breast cancer.3 4 However, once diagnosed, over 97% are treated according to current guidelines with a combination of surgery, radiation and endocrine therapy—treatments similar to those recommended for patients with invasive breast cancer.
The term ‘overdiagnosis’ has been used to define conditions that look like early cancer, but are not destined to cause symptoms or death.5 In 2013, an independent review commissioned by the Department of Health in the UK established that screening saves lives but also that overdiagnosis exists.6 There is a general consensus that much of the overdiagnosis and overtreatment burden in breast cancer derives from the treatment of DCIS. Currently, almost all DCIS is treated according to guideline concordant care (GCC); of those treated for low-risk DCIS, some patients will not benefit if they never develop invasive breast cancer. One possible approach to GCC for low-risk lesions is active surveillance (AS). Currently, only 3% of women in the USA with DCIS opt for AS. Given that much of the treatment for low-risk DCIS may represent overtreatment, there has been global interest to address whether AS, with intervention only for invasive breast cancer, would be sufficient for those women unlikely to have a future DCIS or invasive breast cancer.
Current gaps in evidence
Current treatment options routinely offered for DCIS include surgery (lumpectomy or mastectomy), radiation and endocrine therapy. These options constitute GCC according to National Comprehensive Cancer Network treatment recommendations.7 Between 1991 and 2010, 23.8% of women diagnosed with DCIS in the USA underwent unilateral mastectomy (4.5% bilateral mastectomy), 43% lumpectomy with radiation and 26.5% lumpectomy without radiation.8 Published UK screening data suggest that in some cases, major surgical ‘cancer’ treatment of low-risk DCIS is unnecessary, inappropriate and misleading for the recipient.9 In those women who undergo surgical treatment for DCIS, there may be both short-term and long-term morbidities, including poor cosmesis and the risk of developing persistent pain at the surgical site, with estimates ranging from 25% to 68%.10–13 In addition, patients may experience complications from radiation (cardiac or pulmonary symptoms, secondary malignancies) or reconstruction (infection, loss of implant, need for multiple surgeries). To date, among the 97% of women with DCIS treated with GCC, neither randomised trials nor observational studies have shown a survival advantage of any one treatment option over another.14 Moreover, none of the treatments has ever been compared in a rigorous fashion to AS. The COMET (Comparison of Operative versus Monitoring and Endocrine Therapy) trial for low-risk DCIS is a 5-year phase III, randomised controlled clinical trial that commenced on 1 July 2016. The study was designed with a specific objective: to determine the risks and benefits of GCC compared with those of AS for low-risk DCIS. This protocol is based on version 5 (dated 11 July 2018), approved by QUORUM Centralised Institutional Review Board (CIRB) and all local institutional review boards (IRBs) where relevant. Any future protocol amendments will be submitted to Quorum CIRB or local IRBs, in accordance with institutional requirements, via the sponsor of the study (Alliance Foundation Trials).
Methods and analysis
Trial design and setting
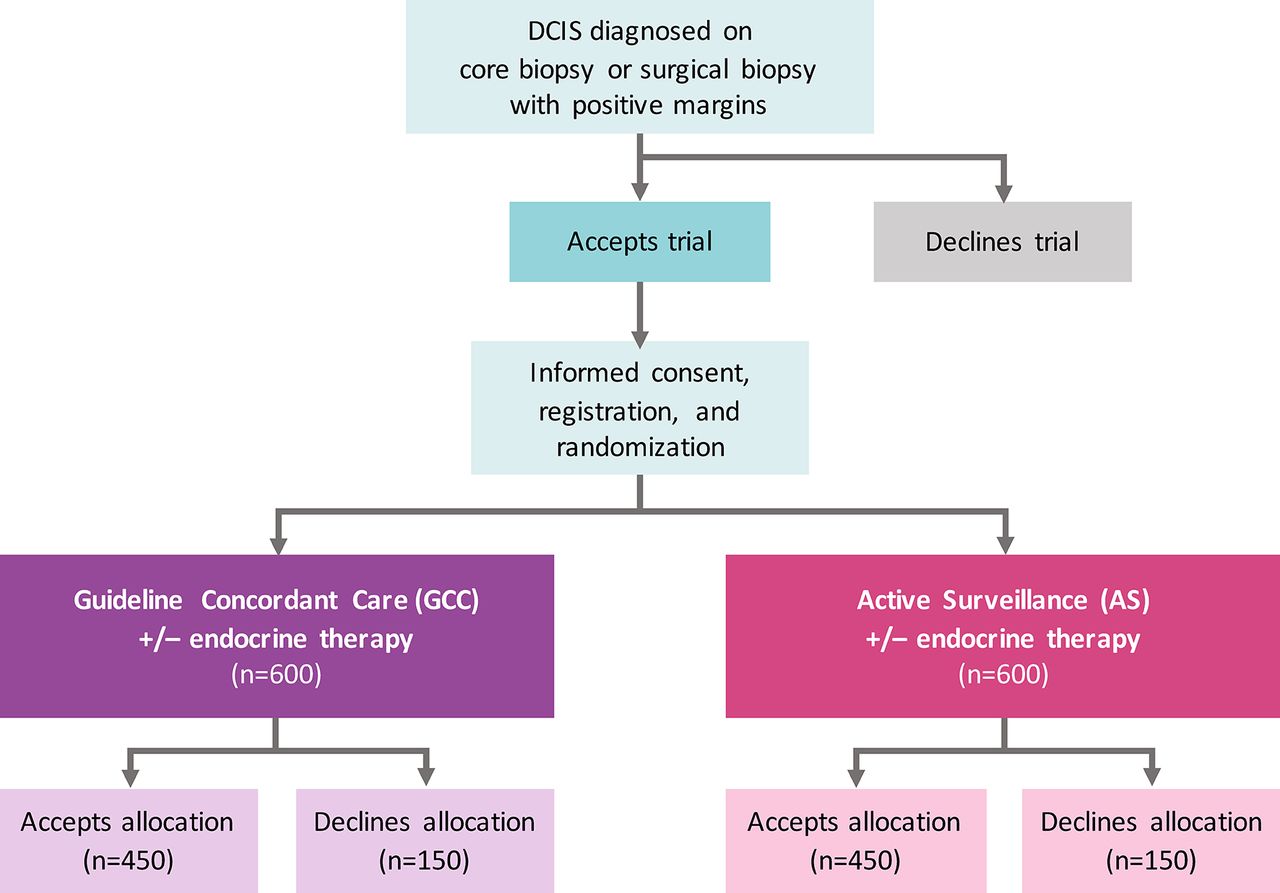
COMET is a phase III randomised controlled clinical trial for low-risk DCIS (figure 1) with two comparator arms, GCC and AS. The study, funded by the Patient-Centered Outcomes Research Institute, is conducted through the Alliance for Clinical Trials in Oncology cooperative group network with plans to open at up to 100 sites in the USA (a list of currently activated sites can be found at ClinicalTrials.gov—NCT02926911). Patients with a new diagnosis of DCIS are identified at participating Alliance study sites and screened for eligibility. Written informed consent is obtained prior to randomisation by site staff, including consent for the potential use of biological specimens in future studies (online supplementary appendix 1). Alliance has obtained a Certificate of Confidentiality from the Department of Health and Human Services in order to protect the privacy of individuals who are subjects of research by withholding their names and other identifying characteristics from all persons not connected with the conduct of Alliance research.
Supplementary file 1

COMET trial schema. Patient flow for accrual and registration. Eligibility criteria for low-risk DCIS include 40 years of age or older, grade I/II DCIS without invasive breast cancer diagnosed on core, vacuum-assisted or surgical biopsy; ER(+) and/or PR(+); HER2(−); and no mass on physical examination or imaging with exception of fibroadenoma at a distinct/separate site from the site of DCIS. The primary study endpoint on which the sample size is based is rate of 2-year invasive breast cancer diagnosis among patients randomised to GCC compared with AS. ITT analyses adjusted for drop-out, non-compliance and contamination will be performed on all randomised patients including those who do and do not accept the arm to which they are randomised. Patient-reported outcome surveys will be collected from all patients who are registered for the study, including those who crossover. Mammograms will be performed q6 months for the index breast and q12 months for the contralateral breast in the AS arm and q12 months in both the index and contralateral breast in the GCC arm. No chest wall imaging will be performed if mastectomy has been performed. AS, active surveillance; COMET, Comparison of Operative versus Monitoring and Endocrine Therapy; DCIS, ductal carcinoma in situ; ER(+), oestrogen receptor positive; GCC, guideline concordant care; HER2 (−), human epidermal growth factor 2 negative; ITT, intention to treat; PR(+), progesterone receptor positive; q, every.
Data collection activities are embedded within the Alliance Statistics and Data Center infrastructure. Resource and data management for the trial follow the established Alliance standard operating processes for the collection, storage and analysis of online case report forms and other data. These procedures include all quality assurance processes that are in place for Alliance clinical trials as well as the use of Medidata Rave as the electronic data capture tool.
Eligibility criteria
Inclusion criteria for COMET were designed to select a group of English-speaking and Spanish-speaking patients at low risk for invasive breast cancer progression based on retrospective epidemiological data. Low-risk criteria were identified for clinical, radiological and pathological features. As a pragmatic trial, central review of imaging and pathology is not performed in real time, but reviewed post hoc. However, given the known limited inter-reviewer correlation between pathologists in the diagnosis of DCIS, the inclusion criteria require that at least two pathologists deem that the histological features meet COMET pathology eligibility criteria. A complete list of COMET inclusion and exclusion criteria are presented in table 1.
Eligibility criteria for the COMET trial
Allocation and randomisation
Allocation and randomisation is conducted by Alliance site staff. Randomisation is computer-generated via Medidata Rave and stratified based on the following factors: age at diagnosis: <55, 55–65, >65; maximum diameter of microcalcifications: <2 cm, 2–5 cm, >5 cm and DCIS nuclear grade: I or II. We record whether the patient has had prior surgical excision for the index diagnosis; this variable will be used for subset analysis but will not be a stratification factor.
Use of endocrine therapy is permitted in both arms with adherence and duration of therapy recorded. Although women are not recruited to the COMET trial if they do not agree to randomisation, those women who are consented but then decline randomisation (or their allocated arm) are still eligible to continue participation in the study if they agree to provide follow-up and survey data. The demographics of the cohort declining their allocated arm will be compared with those of women who adhere to the randomisation. It is anticipated that these data will provide valuable information about the potential for selection bias and will ultimately enable the study to be made more generalisable.
Both intent-to-treat and per-protocol analyses can be biased in the presence of drop-out and non-compliance.15 16 Thus, we intend to complete both of these analyses as sensitivity analyses, but the primary analysis approach will be based on an estimate of the treatment effect among those who comply with arm allocation.17
Study arms
Guideline concordant care
Surgery
Patients randomised to the GCC arm will undergo appropriate surgery for DCIS according to local guidelines. It is expected that patients will complete definitive surgery within 60 days of randomisation. Data on all related surgical procedures, including data on immediate or delayed breast reconstruction, will be collected. If a patient randomised to the GCC arm opts for AS, they will be considered as a ‘crossover’ and will continue to participate in completion of patient-reported outcome surveys.
Radiotherapy
The recommendation for post-surgical radiotherapy should be decided following surgery and recommended according to standard local protocols. The use of post-surgical radiotherapy is not mandated within the trial. However, data pertaining to the use of radiotherapy will be collected.
Active surveillance
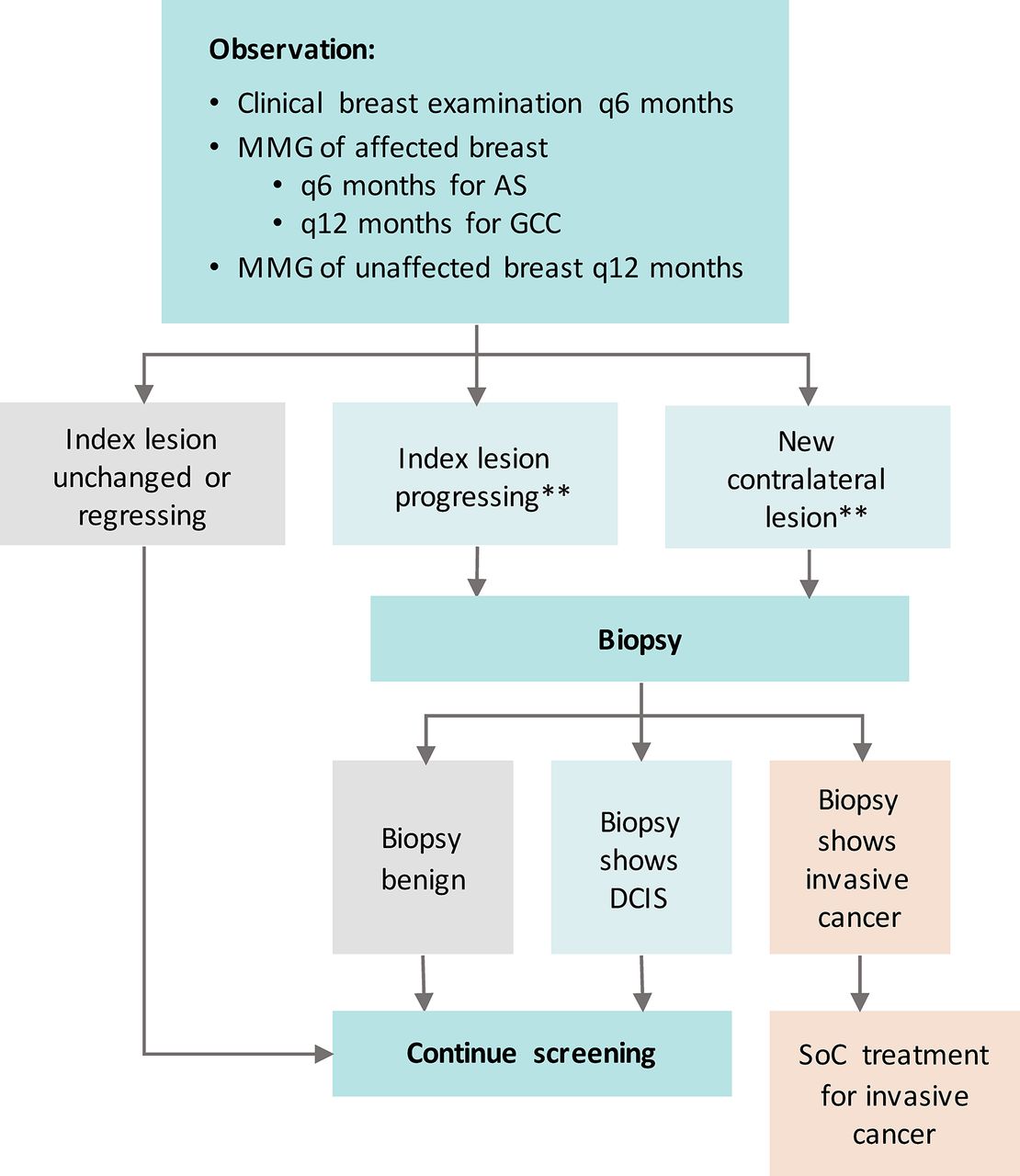
Patients in the AS arm will not undergo surgery unless a biopsy during surveillance documents invasive breast disease which requires surgical intervention. If the patient opts for surgery in the absence of invasive breast cancer, they will be considered as a ‘crossover’ and will continue to participate in completion of patient-reported outcome surveys. Figure 2 presents the surveillance protocol for patients on the AS arm of the study.
{kind=link}
{kind=link}
Surveillance protocol for COMET trial. Mammogram (MMG) not required if mastectomy performed. **Criteria for progression: (A) New mass/architectural distortion/density on surveillance MMG in either breast according to American College of Radiology Breast Imaging Reporting and Data System; (B) Increase in extent of calcifications ≥5 mm in at least one dimension compared with the most recent prior MMG of the index breast; (C) New suspicious findings on other radiological studies (ultrasound, MRI) in either breast. AS, active surveillance; COMET, Comparison of Operative versus Monitoring and Endocrine Therapy; DCIS, ductal carcinoma in situ; GCC, guideline concordant care; SoC, standard of care.
Endocrine therapy
The use of endocrine therapy is not mandatory, but patients are encouraged to discuss this with their providers in both arms of the trial. Selection of endocrine therapy will be determined based on provider recommendation and patient preference, and administered for a maximum duration of 5 years. If applicable, data regarding the use of endocrine therapy (type, duration, adherence and side effects) will be captured at each visit and patient-reported adherence will be measured in follow-up surveys.
All additional follow-up and monitoring beyond that required per protocol (described below) will be conducted according to the standard of care from each provider and institution. The provider will also exercise their best clinical judgement regarding the necessity for baseline laboratory testing (eg, liver function tests, triglycerides) and imaging (eg, breast MRI, dual-energy X-ray absorptiometry scanning).
Surveillance protocol
For both the GCC and AS groups, required surveillance consists of clinical examination, including history and physical examination, every 6 months for a minimum of 5 years and every 12 months thereafter, up to 7 years from the time of registration. Patients on the GCC arm who have not had a mastectomy will have bilateral mammography annually; those on the AS arm will have ipsilateral mammography every 6 months and contralateral mammography every 12 months (table 2).
Schedule of eligibility screening and clinical follow-up
Clinical criteria requiring further investigation include: new breast signs and symptoms such as new breast mass; nipple/skin retraction; nipple discharge and breast oedema/erythema on clinical examination in either breast. Radiographic criteria for biopsy include an increase in extent of calcifications ≥5 mm in at least one dimension compared with the most recent prior MMG in the index breast as well as new suspicious findings on other radiological studies (US, MRI) in either breast (box 1).
Criteria for potential ductal carcinoma in situ progression and indications for biopsy
Clinical criteria
New breast mass on clinical examination in either breast.
Other new breast signs including nipple/skin retraction, nipple discharge, breast oedema/erythema in either breast.
Radiographic criteria
New mass/architectural distortion/density on surveillance mammogram (MMG) in either breast according to American College of Radiology Breast Imaging Reporting and Data System for mammography in assessment of masses and calcifications.31
Increase in extent of calcifications ≥5 mm in at least one dimension compared with the most recent prior MMG in the index breast.
New suspicious findings on other radiological studies (ultrasound, MRI) in either breast.
Duration of follow-Up
Progression, recurrence, new primary disease, residual DCIS (or an additional DCIS lesion) and mortality status will be collected up to 10 years from randomisation.
Outcomes
Endpoints were selected in two broad categories: (1) Clinical outcomes defined as those disease-related and treatment-related outcomes to be collected by research staff from primary source documentation and (2) Patient reported outcomes (PRO) which include an array of relevant quality of life and psychosocial outcomes collected from patient surveys (table 3).
COMET trial primary and secondary endpoints
Primary clinical outcomes
Ipsilateral breast events
Investigational biopsies will be performed in both study arms for suspicion of a new DCIS or invasive breast cancer in the ipsilateral breast as deemed clinically appropriate by the patient’s treatment team. The resulting pathology slides from the biopsy will be reviewed by two pathologists and disease management recommended according to the histological diagnosis. In the GCC arm, any diagnosis will be managed according to standard of care for a local breast event or benign biopsy. In the AS arm, only an invasive breast cancer diagnosis will prompt intervention, according to standard management options.
Contralateral breast events
For both AS and GCC arms, contralateral findings for suspicion of new DCIS or invasive breast cancer will also be managed as deemed clinically appropriate by the patient’s treatment team. The resulting diagnosis will be managed according to best standard practice as determined by provider recommendation and patient preference. If the new contralateral diagnosis is a DCIS that fulfils criteria for the COMET study, the patient can be offered AS or GCC for this diagnosis.
Secondary outcomes
Additional clinical outcomes which are also relevant to the differences between the GCC and AS groups include further surgical procedures and regional or distant metastatic breast cancer events. Since DCIS cells remain trapped within the breast duct and therefore have little potential to spread to distant organ sites and cause symptoms or death, few metastatic events are anticipated.
Patient-reported outcomes
PRO that are potentially important and relevant to women with DCIS will be elicited longitudinally at pre-specified time points during the study (table 4).
Schedule of PRO surveys
Domains including validated measures specific to arm and breast symptoms, body image and decision-making will be collected. To ensure that there is no excessive burden to patients, and to test content flow and clarity, all surveys were piloted by the Patient Leadership Team (PLT) prior to trial initiation. The surveys are provided in print, online or phone interview versions according to patient preference and are also available in Spanish. All PRO data are entered into PRO-CORE, a study-specific survey data collection platform for web-based assessment of PRO, built and managed by the University of North Carolina Patient-Reported Outcomes Core Facility (UNC PRO-Core).
Collaborating sites send MMG files consisting of the last screening and diagnostic MMG studies that immediately predate the diagnostic core/vacuum-assisted biopsy or surgical excision; submission of biospecimens is also a required component of COMET and an integrated part of the consent process (online supplementary appendix 2).
Supplementary file 2
Statistical considerations
Sample size
Sample size for this study was estimated using a two-group test of non-inferiority of proportions, with the 2-year invasive breast cancer rate in the GCC group assumed to be 0.10 based on published studies.18 19 The non-inferiority margin assumed was 0.05 as this was thought to be a clinically meaningful difference between the two arms, beyond which AS could not be reasonably considered to be equivalent to GCC. Based on a one-sided unpooled z-test, with alpha=0.05, a sample size of n=446 per group will have 80% power20 to detect the specified non-inferiority margin. A secondary time-to-event analysis will also be performed.
Planned analysis for clinical outcome data
The primary analysis will not simply follow the intent-to-treat principle but will analyse as randomised all patients with outcomes measured. However, we believe that this trial, as in other studies which randomise to operative versus non-operative arms, will have both non-compliers and contamination, due to patients who will have a desire to avoid surgery as well as patients who, conversely, will have a desire to have any pre-cancerous lesion removed. Thus, the final study design will include a per-protocol component as well as a pragmatic component for those patients who are randomised and decline participation in the assigned arm. We will define a crossover from AS to GCC as any breast surgery on the affected breast in the absence of invasive breast cancer when randomised to AS. Similarly, a crossover from GCC to AS occurs if the patient refuses surgery when randomised to GCC.
Planned subgroup analyses
Although endocrine therapy is not required on either study arm, we will collect data on its use to determine whether it impacts rates of invasive breast cancer in either group.21 Thus, a planned subset analysis of endocrine therapy use will be completed using multivariable logistic regression, with similar adjustments for drop-out and non-adherence. Additionally, factors that may impact the selection of endocrine therapy in both arms such as age and pathological features will also be included. Similarly, we are interested in understanding how imaging modality used may impact assessment of invasive breast cancer during AS, that is, whether MRI detects higher rates of invasive breast cancer than MMG for those patients whose providers opt to include MRI for surveillance. This will also be assessed in the AS group with logistic regression, controlling for factors that could impact selection of MMG versus MRI, such as patient age or breast density. We will also consider menopausal status and baseline risk of breast cancer, and whether these factors influence rate of invasive breast cancer at 2 years. While the study is not powered on these endpoints, these factors are likely to impact outcomes, and thus will be evaluated in planned subset analyses.
Patient and public involvement
Patients, patient advocates and other stakeholders have been actively engaged in the development of this proposal, including the preliminary studies conducted to inform the work. In order to facilitate advocate engagement, we established the PLT. The input and activities of the PLT and advocacy networks guide direction of the trial throughout its entirety.
The PLT have been heavily engaged in the conception of the study (including the original research question) and have partnered in all phases of planning. The PLT collaborated with investigators in the definition of study comparators and outcomes, key constructs to be measured and choice of validated measures to assess those key constructs. The identification of outcomes that the DCIS population of interest notice and care about is particularly relevant in order to provide practical information that can help patients make informed decisions about their health and healthcare. The appropriateness/relevance of survey measures has been reviewed by patient advocates and survey questions have been piloted with them for usability testing.
Patient advocates on the study have diverse involvement or leadership in breast cancer patient advocacy organisations. They have also been strong leaders in the DCIS advocacy community for decades and have deep ties to their constituencies; this will enable them to mentor members of these constituencies who lack this background in order to facilitate their full participation. This active engagement with key stakeholders will be crucial in the compilation of future dissemination strategies/translation of study findings to both professional and patient/public constituencies.
In sum, the PLT: (1) provide input to create effective protocols and survey designs that answer relevant questions for patients, clinicians and research, (2) contribute to the development of educational and implementation tools for clinical sites and patients with DCIS, (3) recruit patient advocates to ‘beta’ test surveys and patient tools, (4) develop and implement strategies to measure the impact of patient involvement on the advancement of engagement science and (5) monitor accrual and participate in the overall implementation of the study.
Ethics and dissemination
The COMET trial will be subject to bi-annual formal review at the Alliance Foundation Data Safety Monitoring Board (DSMB) meetings. At each meeting, the DSMB will review a report of primary and secondary objectives, study schema, definition of primary endpoints, a brief administration summary of current study status, accrual goals versus actual accrual, a summary of patient characteristics to date, a summary of drop-out or crossover from allocated arm, an assessment of data completeness for the various types of data collected, a summary of primary and secondary outcomes by study arm and finally, a summary of adverse events and serious adverse events that will be reported from study entry until 7 years after registration.
Data safety monitoring at interim analysis
Interim analyses for futility/safety will be completed annually, with reporting following Consolidated Standards of Reporting Trials (CONSORT) guidelines for non-inferiority trials.22 We consider stopping under two scenarios: if there is sufficient indication of (1) lack of efficacy or (2) potential harm.
First, we will determine if the deviation of the point estimate of the invasive breast cancer event rate difference for AS versus GCC exceeds 0 by more than 4 SDs or 3 SDs at either of the interim analyses completed when 1⁄2 and 3⁄4 of the expected number of total events has accumulated, respectively. The SD will be computed from the Kaplan-Meier estimates at 2 years. In the case that SD exceeds the predetermined bounds, the probability that AS is non-inferior is minimal, and the trial will be halted due to the lack of efficacy.
Second, the trial was developed based on the premise that the upstaging rate to invasive breast cancer is approximately 10%.23 If this rate is substantially higher than 10%, then we would potentially be exposing AS patients to harm. Thus, if the estimate for the upstaging rate in the GCC arm is significantly greater than 10% based on the Kaplan Meier estimate at 2 years, the trial will be halted due to potential patient harm.
Discussion
Overdiagnosis and overtreatment may be unintended consequences of mammographic screening.24 Given that DCIS is a non-obligate precursor of invasive breast cancer, for those women whose DCIS might never progress even without treatment or whose treatment and outcomes may not differ even if invasion occurs, there is a pressing need to study more selective clinical strategies than the current, non-risk-based therapies for DCIS originally intended for invasive breast cancer. For DCIS at low-risk of progression such as low-grade, small, non-palpable lesions, there may be no significant benefit to surgery or radiation and a de-escalation approach should be tested as it has been in other cancers (eg, prostate cancer).25 26 There is recognition that high-grade DCIS is more likely to progress to an invasive breast cancer and these patients are excluded from the study. Given the lead-time between the development of DCIS and appearance of invasive breast cancer,21 there may also be a case for tailoring intervention by age and presence of competing comorbidities.
Global collaboration
Adoption of significant practice changes in breast cancer treatment has often required consideration of multiple sources of information. Thus, compatibility with other trials is an important goal for the implementation of findings from the COMET study. The LOw Risk dcIS study (LORIS trial; ISRCTN27544579)27 28 is a randomised controlled trial of AS versus GCC in the UK, which opened to accrual in 2015. The patient populations, healthcare environments and the clinical trials organisation of the COMET and LORIS studies represent an exceptional opportunity to combine resources and strategies, to compare outcomes and to identify similarities and differences in DCIS diagnosis, treatment and surveillance policies both from a patient population and a healthcare systems perspective. To that end, LORIS principal investigators have worked with the COMET team in order to closely align the two studies and allow future meta-analysis of both clinical and PRO endpoints. Specifically, we have prospectively designed the eligibility criteria, outcomes and surveillance protocol which, while not identical in every instance, will nevertheless allow for a planned meta-analysis at completion of both studies. In addition, there is a randomised, international, multi-centre, phase III non-inferiority trial being conducted in the Netherlands (The LORD—LOw Risk DCIS study)29 as well as other global efforts to identify biological components of DCIS ‘risk’; for example, the Prevent Ductal Carcinoma In Situ Invasive Overtreatment Now (PRECISION) study.30
The broad, long-term objective of this proposal is to provide high-quality evidence regarding outcomes of treatment versus surveillance for DCIS and to determine whether data support the inclusion of AS in treatment guidelines for DCIS. It is anticipated that the evidence provided by the COMET study, together with data collected from the other low-risk DCIS studies, will enable patients and stakeholders to make better informed decisions about potential management options for low-risk DCIS.
The COMET study represents an important opportunity to address a highly relevant healthcare issue with broad-reaching health, social and economic implications. Moreover, we hope that this study may provide a framework for evidence development in other low-risk conditions where overtreatment is an emerging concern.
Acknowledgments
We gratefully acknowledge all patients and families who are participating in DCIS clinical trials worldwide whose commitment allow for important advances in DCIS.
References
Footnotes
Contributors The PI and first author of this paper (ESH) was instrumental in the compilation of this study protocol. Each co-author (TH, TL, EF, DP, DB, DC, AB, CK, SR, AT, AW, AP) contributed equally to subsequent development of the protocol. EF, DP, DB and DC form the COMET Study Patient Leadership Team.
Funding This work was supported through a Patient-Centered Outcomes Research Institute (PCORI) Award (PCS-1505-30497). The sponsor of the COMET study is Alliance Foundation Trials (https://alliancefoundationtrials.org). Correlative studies are supported by the Breast Cancer Research Foundation (BCRF-17-173). ESH is supported by a Duke Comprehensive Cancer Center Grant (5P30CA014236-44). Patient-reported outcome data are collected via the University of North Carolina Patient Reported Outcomes Core, which is supported in part by grants from the National Institute of Health (DK056350) to the University of North Carolina Nutrition Obesity Research Center, and the US National Cancer Institute (P30 CA016086) to the Lineberger Comprehensive Cancer Center.
Disclaimer All statements in this article are solely those of the authors and do not necessarily represent the views of PCORI, its Board of Governors or Methodology Committee.
Competing interests None declared.
Ethics approval Quorum Centralised Institutional Review Board.
Provenance and peer review Not commissioned; peer reviewed for ethical and funding approval prior to submission.
Patient consent for publication Not required.