Article Text

Abstract
Objective Lesogaberan (AZD3355) is a novel γ-aminobutyric acid B-type receptor agonist designed to treat gastro-oesophageal reflux disease (GERD) by inhibiting transient lower oesophageal sphincter relaxations. A randomised, double-blind, placebo-controlled, multi-centre phase IIb study was performed to assess the efficacy and safety of lesogaberan as an add-on to proton pump inhibitor (PPI) therapy in patients with GERD who are partially responsive to PPI therapy (ClinicalTrials.gov reference: NCT01005251).
Design In total, 661 patients were randomised to receive 4 weeks of placebo or 60, 120, 180 or 240 mg of lesogaberan twice daily, in addition to ongoing PPI therapy. Symptoms were measured using the Reflux Symptom Questionnaire electronic Diary. Response to treatment was defined as having an average of ≥3 additional days per week of not more than mild GERD symptoms during treatment compared with baseline.
Results In the primary analysis, 20.9%, 25.6%, 23.5% and 26.2% of patients responded to the 60, 120, 180 and 240 mg twice daily lesogaberan doses, respectively, and 17.9% responded to placebo. The response to the 240 mg twice daily dose was statistically significantly greater than the response to placebo using a one-sided test at the predefined significance level of p<0.1. However, the absolute increases in the proportions of patients who responded to lesogaberan compared with placebo were low. Lesogaberan was generally well tolerated, although six patients receiving lesogaberan developed reversible elevated alanine transaminase levels.
Conclusions In patients with GERD symptoms partially responsive to PPI therapy, lesogaberan was only marginally superior to placebo in achieving an improvement in symptoms.
- GABAB receptor agonists
- gastro-oesophageal reflux
- lesogaberan
- proton pump inhibitors
- gastro-oesophageal reflux disease
- barretts metaplasia
- barretts carcinoma
- barretts oesophagus
- gastrointestinal endoscopy
Statistics from Altmetric.com
- GABAB receptor agonists
- gastro-oesophageal reflux
- lesogaberan
- proton pump inhibitors
- gastro-oesophageal reflux disease
- barretts metaplasia
- barretts carcinoma
- barretts oesophagus
- gastrointestinal endoscopy
Significance of this study
What is already known on this subject?
-
The symptoms of gastro-oesophageal reflux disease (GERD) are experienced by 10%–20% of the European and North American population.
-
Current treatment is by suppression of gastric acid secretion using proton pump inhibitors (PPIs).
-
Approximately 20%–30% of patients continue to experience residual GERD symptoms, despite PPI therapy.
-
Lesogaberan is a γ-aminobutyric acid B-type receptor agonist that inhibits transient lower oesophageal sphincter relaxations and has been developed for the treatment of GERD as an add-on to PPI therapy in patients who have a partial response to PPI therapy.
What are the new findings?
-
This was a phase IIb study testing the effect of varying doses of lesogaberan in addition to proton pump inhibitor (PPI) therapy on gastro-oesophageal reflux disease symptoms in the target patient population.
-
The highest dose of lesogaberan tested (240 mg twice daily) produced a statistically significant increase in the proportion of patients responding to treatment using a one-sided test at a predefined significance level of p<0.1.
-
However, the absolute increase in the proportion of responders was too small to be considered clinically relevant in the broad population of patients who have a partial response to PPI therapy that was tested.
-
Lesogaberan was generally well tolerated throughout the study, although six patients receiving active treatment developed reversible elevated alanine transaminase levels >5 times the upper limit of normal.
How might it impact on clinical practice in the foreseeable future?
-
The development of lesogaberan as an add-on to proton pump inhibitor (PPI) therapy for the treatment of gastro-oesophageal reflux disease in patients whose symptoms show a partial response to PPI therapy has been halted.
-
More research is required to determine whether reflux inhibition would be of greater benefit in a different patient population.
Introduction
Gastro-oesophageal reflux disease (GERD) affects 10%–20% of the European and North American population and is characterised by symptoms of heartburn and regurgitation.1 Symptoms are thought to be primarily caused by exposure of the oesophagus to stomach acid, and current treatment involves suppression of gastric acid secretion using proton pump inhibitors (PPIs).2 Although most patients experience symptom resolution on PPI treatment, approximately 20%–30% of patients continue to experience residual symptoms.3 The aetiology of symptoms in these patients is poorly understood, but they may be functional or caused by ongoing oesophageal exposure to acidic, weakly acidic or alkaline refluxate.3–6
The lower oesophageal sphincter is the major barrier protecting the oesophagus from constant exposure to stomach contents. However, sphincter pressure is reduced by transient lower oesophageal sphincter relaxations (TLESRs), a physiological phenomenon triggered by distension of the stomach and allowing the evacuation of gas. TLESRs are thought to be responsible for approximately 80% of all reflux episodes in patients with GERD, and are therefore a potential target for treatment in patients who do not fully respond to PPI therapy.7 ,8
Stimulation of γ-aminobutyric acid B-type receptors (GABAB), both peripherally in the stomach and oesophagus and centrally in the brainstem, has been demonstrated to inhibit TLESRs.9 Baclofen, an existing GABAB agonist, has been shown to inhibit both TLESRs and reflux episodes in healthy individuals and patients with GERD,10–12 and to reduce reflux symptoms when used as an add-on to ongoing PPI therapy in patients who have persistent heartburn or regurgitation.13 However, the utility of baclofen in the treatment of GERD is limited because of centrally mediated adverse effects, with up to 10% of patients experiencing tiredness and sleepiness.10 ,12
Lesogaberan (AZD3355) was developed as a peripherally acting selective GABAB agonist with potential utility in the treatment of GERD. The compound is sequestered in central neurons and glial cells, resulting in limited access to receptors in the central nervous system.14 Previous studies have demonstrated that lesogaberan successfully inhibits TLESRs, increases lower oesophageal sphincter pressure and inhibits reflux episodes in both dogs and humans.14–16 Furthermore, the compound has demonstrated a favourable safety and tolerability profile in early clinical trials.15 ,17
A previous phase IIa study with a 65 mg twice daily dose of lesogaberan demonstrated a modest effect in reducing GERD symptoms when used as an add-on to PPI therapy in patients with persistent reflux symptoms despite PPI therapy.17 A randomised, double-blind, placebo-controlled, multi-centre phase IIb study was therefore performed to determine the efficacy and safety of higher doses of the drug (ClinicalTrials.gov reference: NCT01005251).
Methods
Patients
The study was performed at 109 study centres in seven different countries: 68 in the USA, 10 in Canada, nine in France, seven in Germany, six in Hungary, six in Romania and three in Latvia. Men and women aged 18–70 years were eligible for enrolment in the study if they had experienced reflux symptoms for at least 6 months, and if they had ongoing symptoms despite having received a minimum of 4 weeks of individually optimised PPI therapy (treatment that, according to the investigator's judgement, could not be further improved by changing the brand or dosing (timing of administration) of the PPI). Doses were required to be within the approved range for any GERD indication according to the country or region label. Twice daily PPI dosing was not allowed, and patients who had been endoscopically diagnosed with reflux oesophagitis in the 8 weeks before enrolment were required to have received a minimum of 8 weeks of PPI therapy. Ongoing symptoms were defined as ≥3 days with at least moderate burning feeling behind the breastbone (heartburn) and/or ≥3 days with at least moderate unpleasant movement of material upwards from the stomach (regurgitation) in the week before enrolment, reported using an electronic 7-day recall questionnaire (the Reflux Symptom Questionnaire (RESQ)).18 All patients who had not undergone an endoscopy in the previous 24 months, or whose most recent endoscopy in the previous 24 months had shown oesophageal mucosal breaks, underwent an endoscopy at baseline.
The main exclusion criteria included having a body mass index >35.0 or <18.5 kg/m2 (in order to limit the variability in mg/kg dosing between patients), clinically significant disorders that could compromise safety during the study (cardiovascular, respiratory, hepatic, renal, metabolic or neurological disorders, or gastrointestinal disorders besides GERD), or a history of syncope, heart disease, malignant disease, electrolyte imbalances or severe allergic or hypersensitivity reactions. Patients were also excluded if they had shown no improvement at all in their reflux symptoms during PPI therapy or if they were using concomitant drugs that could potentially interfere with the pharmacodynamic effects of lesogaberan (such as baclofen or supplements containing GABA), alter gastrointestinal symptoms (such as type-2 histamine receptor agonists) or cause damage to the mucosal lining of the gastrointestinal tract (such as non-steroidal anti-inflammatory drugs or acetylsalicylic acid >162 mg/day). Women were excluded if they were pregnant or breast feeding, and women of childbearing potential were required to use a highly effective contraceptive method.
All patients provided informed consent before the initiation of any study-specific procedures, and the study was carried out in accordance with the Declaration of Helsinki. A three-member Data Safety and Monitoring Board with no affiliation to the sponsor or other involvement in the trial assessed the interim safety data. Reporting of this trial complies with the CONSORT Group's statement for transparency in clinical trials.19
Study design
An outline of the study design is shown in figure 1. Following enrolment, patients entered an 8–26-day screening phase during which they continued to receive their ongoing PPI therapy and recorded their symptoms twice daily using the RESQ electronic Diary (RESQ-eD).20 Patients were eligible for randomisation if they recorded ≥3 days with a burning feeling behind the breastbone and/or unpleasant movement of material upwards from the stomach of at least moderate intensity in the final 7 days of screening. Eligible patients were randomised in equal proportions to receive placebo or 60, 120, 180 or 240 mg doses of lesogaberan twice daily, in addition to continuing PPI therapy.
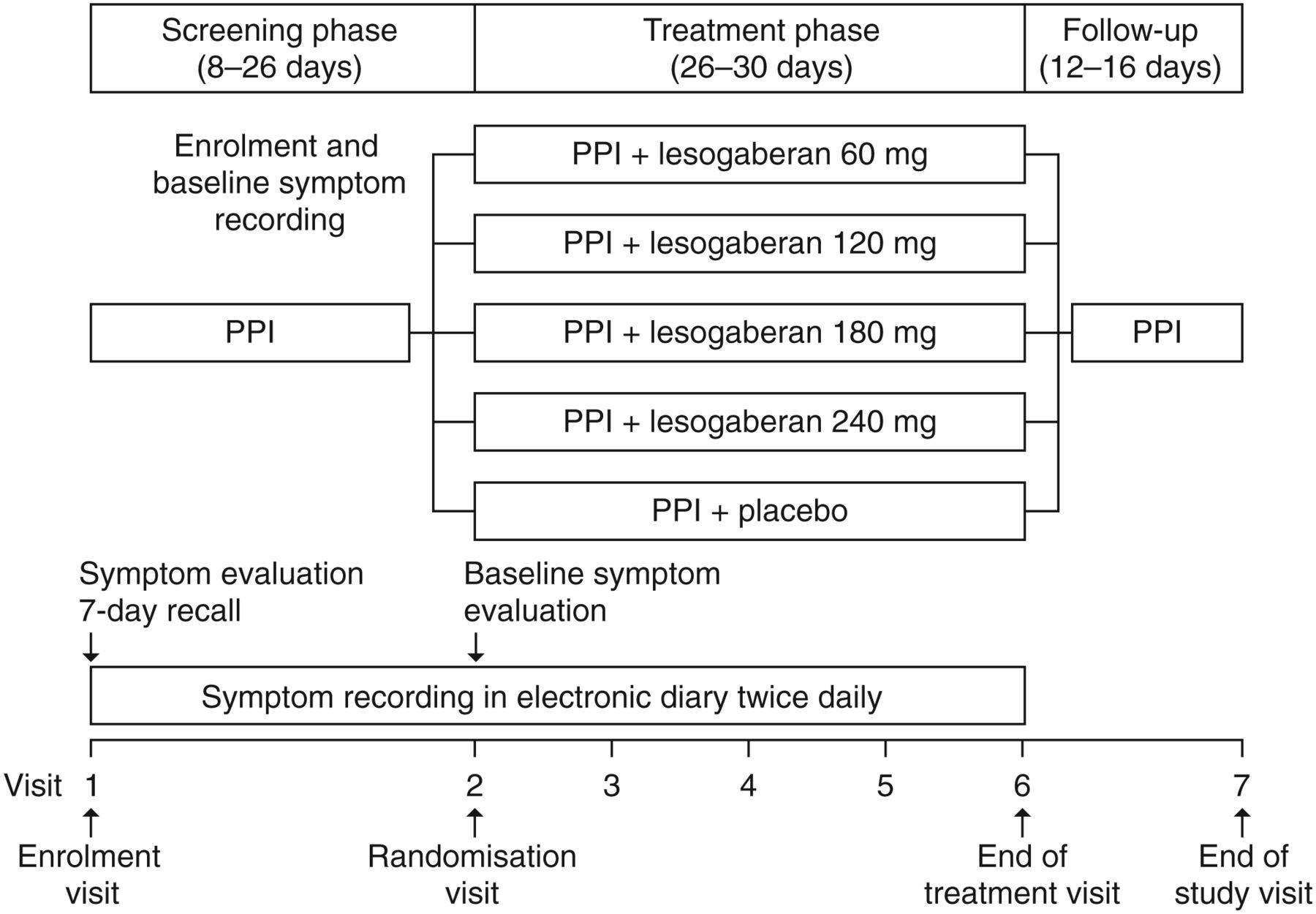
Study design. Endoscopies were carried out if applicable according to the protocol. Lesogaberan doses were given twice daily. PPI, proton pump inhibitor.
Patients were randomised to treatment groups sequentially, based on a balanced block randomisation schedule. All patients, investigators and study personnel were masked to patient randomisation. The treatment phase lasted for 4 weeks (range 26–30 days), during which patients were instructed to take the study medication 30 min before breakfast and 30 min before their main evening meal, and continued to record their symptoms twice daily using the RESQ-eD. Patients attended weekly study centre visits during the treatment phase of the trial and had a follow-up visit 2 weeks after the treatment phase ended. During study centre visits, patients underwent safety monitoring and a review of RESQ-eD compliance. Patients were discontinued from the study if they met prespecified safety outcomes described below.
Study assessments
The RESQ-eD is a patient-reported outcomes measure that has been previously demonstrated to be valid, reliable and responsive to change in patients who have a partial response to PPI therapy.20 The questionnaire consists of 13 items in which patients indicate symptom intensity on a 6-point Likert scale, graded from ‘did not have’ to ‘severe’. These items combine into an Overall symptoms domain and four separate symptom domains:
-
Heartburn
-
Regurgitation
-
Hoarseness, cough, difficulty swallowing
-
Burping.
Response to treatment was defined as experiencing ≥3 additional days on average per week with not more than mild Overall symptoms (ie, reporting not more than mild intensity symptoms in any of the 13 items) during treatment compared with baseline (the final 7 days of screening). The change from baseline to the treatment period in the proportion of days with not more than mild symptoms was also assessed for each separate symptom domain of the RESQ-eD. The change in the proportion of days with not more than mild Overall symptoms was used to estimate an Emax dose–response curve. Analysis was for the full analysis set: all those who received one or more doses of the treatment option to which they were randomised.
Safety measurements
At each study centre visit, patients reported any adverse events (AEs) experienced and provided blood samples for laboratory analysis. Patients also underwent blood pressure and pulse rate measurements, orthostatic tests and digital ECG. An orthostatic blood pressure reaction was defined as a reduction in systolic blood pressure of ≥20 mm Hg or a reduction in diastolic blood pressure of ≥10 mm Hg within 1 min of standing. Any patients who spontaneously reported symptoms of paraesthesia underwent further questioning to determine the severity, frequency and duration of the symptoms. In addition, all patients were questioned for symptoms of syncope, faintness or dizziness. Discontinuation criteria were: daily episodes of paraesthesia for ≥7 consecutive days; syncopal episodes experienced due to any cause; a presyncopal episode described by the patient as severe; a corrected QT interval of ≥500 ms; or serum alanine transaminase (ALT)/aspartate transaminase levels either ≥5 times the upper limit of normal (ULN) or ≥3 times the ULN with serum bilirubin ≥2 times the ULN or clinical signs or symptoms indicating liver dysfunction.
Sample size determination
When the responder definition was applied retrospectively to data from a clinical trial that included a similar patient population (RESQ-eD validation study, ClinicalTrials.gov reference: NCT00703534),20 the proportion of responders was 24% in the placebo group (data on file). With 130 patients in each treatment group, assuming a true placebo response rate of 24%, a one-sided χ2 test at a predefined significance level of 10% would identify an increase of 15 percentage points (ie, a response to treatment of at least 39%) with an approximate power of 90%. An observed difference of at least 8% would therefore be statistically significant at the 10% level. Based on these calculations, 130 patients per treatment group were sufficient to address the primary objective. Sample size calculations were made using nQuery V.4.0 (Statistical Solutions, Cork, Ireland).
Statistical analyses
ORs, CIs and p values for the response to each dose of lesogaberan compared with placebo were calculated using a logistic regression model, with the binary response outcome as a response variable, dose as a factor variable and the proportion of days with not more than mild symptoms at baseline as a covariate. For the primary endpoint analysis (proportion of patients responding to treatment), significance was defined as p<0.1 using a one-sided test. This level of significance was chosen as an acceptable level of continuation risk, that is, a 10% risk of continuing into phase III with an ineffective drug. A two-sided 80% CI is presented, corresponding to a one-sided test at a 10% significance level. Multiplicity of testing was addressed by using a step-down procedure, starting by testing the significance of the response to the highest dose of lesogaberan and continuing to lower doses sequentially only if the null hypothesis of no difference between the response to lesogaberan treatment and placebo could be rejected. ORs, 95% CIs and p values for the change in the proportion of days with not more than mild symptoms in each separate domain of the RESQ-eD during treatment compared with baseline were calculated by pairwise comparison with placebo using logistic regression (with cumulative logits and a proportional odds model) and a one-sided Wilcoxon rank-sum test (not adjusted for multiplicity), respectively.
The Emax dose–response curve was estimated using the following formula: where Y is the response to lesogaberan, E0 is the response to placebo, Emax is the maximum achievable increase above the placebo response, ED50 is the dose that produces 50% of the Emax effect, D is the dose level and β is a sensitivity measure of the response variable with respect to relative increases in dose. Statistical analyses were performed using SAS® V.8.2 (Cary, North Carolina, USA).
where Y is the response to lesogaberan, E0 is the response to placebo, Emax is the maximum achievable increase above the placebo response, ED50 is the dose that produces 50% of the Emax effect, D is the dose level and β is a sensitivity measure of the response variable with respect to relative increases in dose. Statistical analyses were performed using SAS® V.8.2 (Cary, North Carolina, USA).
Results
Patient disposition and baseline characteristics
In total, 1471 patients were enrolled in the study, of whom 661 were eligible for randomisation (figure 2). Of the randomised patients, 580 completed treatment, with the most common reasons for discontinuation being AEs (25 patients of whom four were in the placebo group, no lesogaberan dose association) and patient decision (22 patients of whom nine were in the placebo group, no lesogaberan dose association). The mean age of randomised patients was 47.7 years, and fewer men were included than women (43% vs 57%) (table 1). Most patients (84.6%) were White subjects, with an average body mass index of 27.9 kg/m2 and an average GERD history of 8.9 years. The most commonly used PPI was omeprazole (42.8% overall), followed by esomeprazole, lansoprazole, pantoprazole and rabeprazole. A total of 89.6% of patients had been taking their PPI for more than 2 months before the start of the study. There was a very high symptom burden among randomised patients, with 75.5% of patients experiencing symptoms of more than mild intensity on a daily basis and 95.7% experiencing symptoms of more than mild intensity on ≥5 days per week (table 2).

Study flow diagram. Lesogaberan doses were given twice daily. AE, adverse event.
Patients' baseline demographics and characteristics (randomised patients)
Number of days with symptoms of more than mild intensity at baseline (randomised patients)
Primary analysis
Overall, the proportions of patients who responded to the 60, 120, 180 and 240 mg twice daily doses of lesogaberan were 20.9%, 25.6%, 23.5% and 26.2%, respectively, while 17.9% of patients responded to placebo (figure 3). The 240 mg twice daily dose of lesogaberan was shown to be associated with a significantly greater response than placebo according to the predefined significance level of p<0.1 (OR, 1.63; 80% CI 1.11 to 2.41). However, the effect of the 180 mg twice daily dose of lesogaberan was not significant (p=0.1213). The significance of the 120 mg and 60 mg twice daily doses was therefore not tested, in accordance with the step-down procedure.

Response to treatment according to the primary responder definition. Lesogaberan doses were given twice daily. *One-sided p<0.1, relative to placebo, derived from logistic regression analysis. Note that the significance of the 60 mg and 120 mg twice daily doses of lesogaberan was not tested since the 180 mg twice daily dose of lesogaberan showed p≥0.1.
Secondary analysis
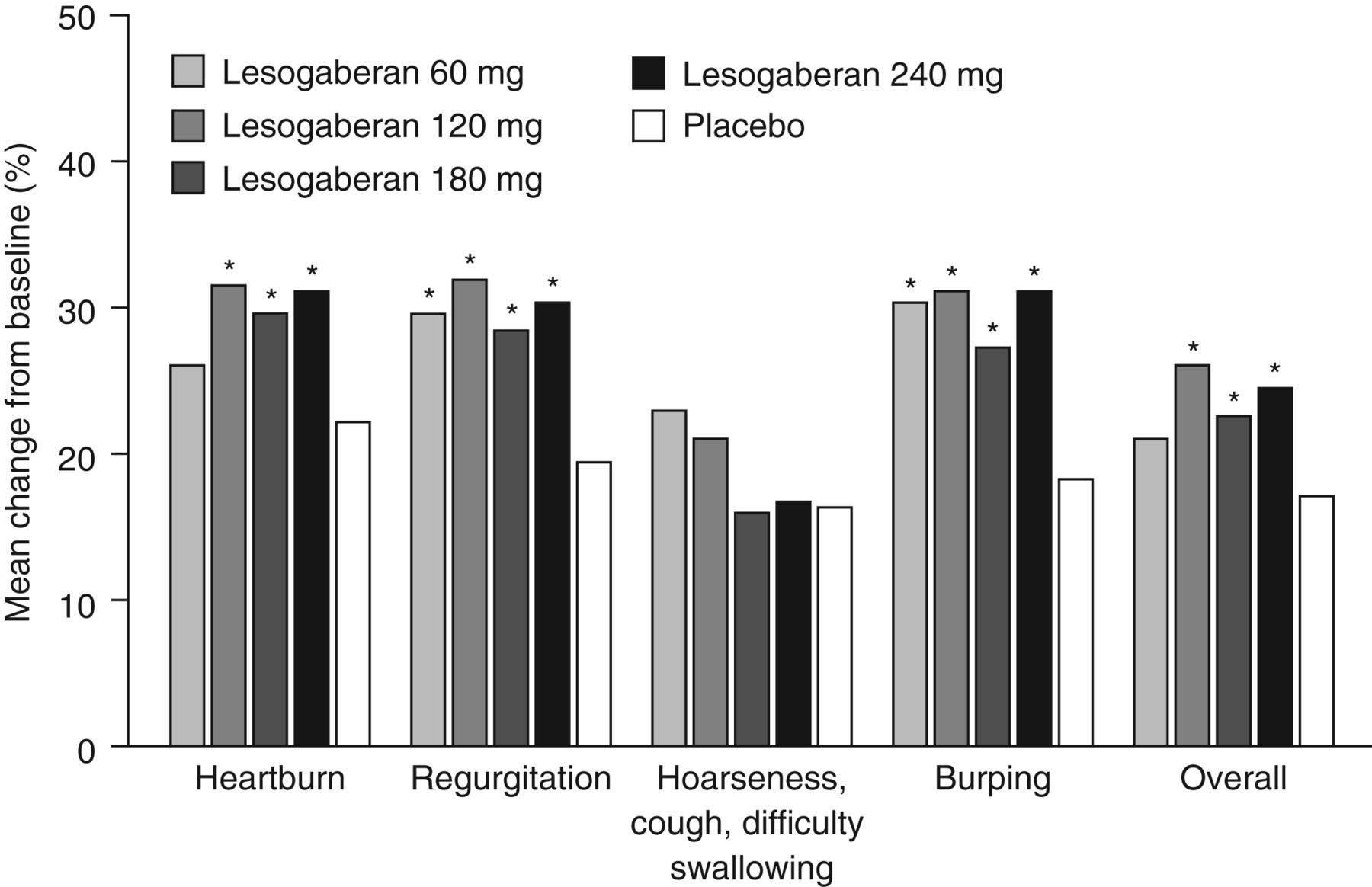
The proportions of days with not more than mild Regurgitation and Burping during treatment compared with baseline were increased relative to placebo for all doses of lesogaberan tested (p<0.05), and the proportions of days with not more than mild Heartburn and Overall symptoms during treatment compared with baseline were increased relative to placebo for all doses of lesogaberan ≥120 mg (p<0.05) (figure 4). However, compared with placebo these increases corresponded to an average of <1 additional day per week with not more than mild symptoms, relative to baseline. No dose of lesogaberan produced a significant increase in the proportion of days with not more than mild Hoarseness, cough, difficulty swallowing during treatment relative to baseline. The Emax dose–response curve estimate reached a plateau at the 120 mg twice daily dose of lesogaberan (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change from baseline in the proportion of days with not more than mild symptoms during treatment. Lesogaberan doses were given twice daily. *One-sided p<0.05 (Wilcoxon rank-sum test) relative to placebo.
Safety analysis
AEs were reported by 37.0%, 38.7%, 44.1% and 46.0% of patients taking the 60, 120, 180 and 240 mg twice daily doses of lesogaberan, respectively, and by 30.7% of patients receiving placebo (table 3). The most common AEs were paraesthesia, diarrhoea, pruritus, dizziness and nausea. Diarrhoea and pruritus were notably more common in the lesogaberan treatment groups than in the placebo group.
Number of patients who had at least one AE during the treatment period, and abnormal laboratory test results at any time postbaseline
Only one patient experienced a serious AE (metastatic gastric cancer) during the active treatment phase, and this patient was in the placebo group. One patient in the 240 mg twice daily lesogaberan treatment group experienced a serious AE (non-cardiac chest pain) during follow-up, but this was not considered to be causally related to the study medication. No patients died during the study. There were no clinically relevant increases in paraesthesia symptoms or syncope/faintness/dizziness in the lesogaberan treatment groups compared with the placebo group.
No patients experienced an episode of syncope during the study, although one patient receiving the 60 mg twice daily dose of lesogaberan experienced an episode of presyncope. There were no clinically important trends in mean laboratory values or changes over time in orthostatic pulse or blood pressure (data not shown). A slight increase in the frequency of orthostatic blood pressure reactions was observed at the highest dose of lesogaberan, although no patients experienced clinical symptoms during the test.
Six patients taking lesogaberan developed ALT levels >5 times the ULN during the study. There was no obvious lesogaberan dose association, and changes were reversible. In two patients, possible causes for the elevated liver transaminases other than the study medication were noted. One patient had ultrasound findings consistent with gallbladder disease, and associated clinical symptoms including anorexia, abdominal pain, nausea and vomiting. In this patient, elevated ALT levels were first detected 2 weeks after treatment ended, bilirubin levels were >2 times the ULN and alkaline phosphatase levels were also raised. The second patient had started ibuprofen 1600 mg/day and hydrocodone/paracetamol the week before the elevated liver transaminases were detected. However, in the remaining four patients no explanation for elevated liver transaminases other than the study medication could be established.
Discussion
The results presented here show a modest treatment effect of lesogaberan on reflux symptoms when used as an add-on to PPI therapy in patients who have a partial response to PPIs. With the responder definition of patients experiencing ≥3 additional days on average per week of not more than mild GERD symptoms during treatment compared with baseline, using the step-down approach, only the highest dose of lesogaberan (240 mg twice daily) was demonstrated to produce a statistically significant response at the 10% significance level. However, the Emax dose–response curve showed a plateau in the response to lesogaberan at the 120 mg twice daily dose, making it unlikely that the results were due to a failure to test high enough doses to observe an optimum response.
The absolute increase in the proportion of responders was small, with 8.3% more patients responding to the 240 mg twice daily lesogaberan dose than to placebo. The corresponding number needed to treat is 12.0. Given that most patients with GERD symptoms partially responsive to PPIs have non-erosive disease, and that any additional medication to augment the effect of PPIs would presumably need to be taken long term, this degree of effectiveness is unlikely to be sufficient to make lesogaberan treatment a consideration for most physicians. In the separate symptom domains of the RESQ-eD that showed a significant response to treatment (Heartburn, Regurgitation, Burping and Overall symptoms), changes from baseline in the proportion of days with not more than mild symptoms corresponded to an average gain of <1 extra day per week compared with placebo.
Lesogaberan was generally well tolerated throughout the study, although six patients receiving active treatment developed elevated ALT levels >5 times the ULN. In four of these patients, no causes for changes in liver transaminase levels other than study medication were noted. There were few central nervous system mediated AEs, which supports the hypothesised peripheral mode of action of the drug.14
An earlier phase IIa study demonstrated that in a similar population of patients who have a partial response to PPIs, a 65 mg twice daily dose of lesogaberan in addition to PPI therapy significantly increased the proportion of responders relative to placebo.17 Using a slightly different responder definition than the present work, the increase in the proportion of responders relative to placebo was similarly modest (16% vs 8%; one-sided p=0.026).17 At the 65 mg dose, a 25% inhibition of TLESRs and a 35% inhibition of reflux episodes were observed in patients in a pharmacodynamic study.15 Dose-finding studies in dogs have shown that 50% inhibition of TLESRs can be achieved using lesogaberan,14 suggesting the potential for greater inhibition of reflux episodes and effects on symptoms in humans at doses higher than 65 mg. The present study was therefore designed to build on these earlier results, testing higher doses of lesogaberan with the aim of determining an optimum dose based on efficacy and safety considerations. However, the maximum therapeutic gain at the substantially higher doses assessed in the current study proved to be similar to that in the earlier phase IIa study.
There are a number of potential reasons for the limited therapeutic effect of lesogaberan seen in this study. Although the included patients were representative of the intended target population for lesogaberan treatment and selected based on persistent symptoms considered typical for GERD, there was no requirement for enrolled patients to have objective evidence of pathological reflux, either acidic or non-acidic, by endoscopy, impedance monitoring, pH-metry or other metrics. There was therefore no clear-cut evidence that symptoms were actually caused by reflux. Dilution of the patient population to an unpredictable extent with individuals who have functional or non-reflux related symptoms unlikely to respond to lesogaberan treatment would make detection of a significant treatment effect among patients who could benefit from the mode of action of lesogaberan less likely. It has been estimated that of all patients with GERD symptoms, up to 20% may be experiencing functional heartburn,21 and this proportion is likely to be higher among patients who have a partial response to PPI therapy.22
Limiting the target population by requiring pathological impedance pH-metry measurements would have identified a group of patients with objective evidence of ongoing reflux, who might therefore be more likely to benefit from reflux inhibition. Currently, no approved treatment options are available for these patients if they do not respond sufficiently to PPI therapy. However, it would have limited the applicability of the trial for the future use of lesogaberan in the clinic, since such testing is not widely available or commonly used. We attempted to decrease the proportion of subjects with functional heartburn in the study group by excluding patients who had experienced no improvement at all while taking PPIs. However, this may not have been sufficient, since a partial response to PPI therapy among patients with functional symptoms may be mediated by a placebo effect. It may also be difficult for patients to determine whether no response at all to therapy has been experienced.
Inclusion of symptoms in the responder definition which have a less well established association with reflux, such as cough and hoarseness, in the broad responder definition may have limited the sensitivity of the primary outcome measure. However, changing the responder definition to exclude symptoms more weakly associated with reflux was not tenable, as these symptoms were identified as relevant to the target patient population in the generation of the patient-reported outcome measure used in this study.20 It is also notable that when symptoms that would be expected to be more firmly associated with TLESRs, such as regurgitation and burping, were analysed separately, the effect of lesogaberan remained small. These symptom domains, however, did show a more positive association with lesogaberan treatment than Heartburn and Hoarseness, cough, difficulty swallowing, showing unadjusted p values of <0.05 at all doses of lesogaberan tested when the number of days with not more than mild symptoms were analysed. Heartburn showed a p value of <0.05 at doses of 120 mg and higher, and Hoarseness, cough, difficulty swallowing did not show a p value of <0.05 at any dose tested. A patient population selected on the basis of the predominance of symptoms of regurgitation and burping may therefore have been more likely to respond to TLESR inhibition and could thus have shown a more positive response to treatment than the broad patient population tested in this study. However, further clinical studies would be required to test this hypothesis.
It is not currently known how much of a reduction in reflux episodes is needed to translate into a clinically relevant effect on symptoms. It is possible that the maximum inhibition of TLESRs attainable by the use of a peripherally acting GABAB agonist may be too small to prevent enough reflux episodes from occurring to impact sufficiently on symptom generation. It is of note that two other reflux inhibitors have also failed to generate a clinically significant treatment response in the target patient population.23 ,24 It seems plausible that patients who have a partial response to PPIs are more sensitive to reflux than those who have a complete response to PPIs, as they may experience symptoms generated by weakly alkaline or weakly acidic refluxate.3–6 A partial inhibition of reflux episodes may therefore be insufficient to prevent symptom generation in these patients. Finally, the mechanism of symptom generation in patients with reflux symptoms partially responsive to PPI therapy is poorly understood. Although we concentrated on TLESR inhibition, if other mechanisms (including hypersensitivity and central nervous system mediated mechanisms) are important in the generation of these symptoms, an agent such as lesogaberan might not be expected to be effective. It also remains possible that lesogaberan could be effective as a monotherapy, either in patients who respond fully to PPI therapy or in those who have a partial response. Although PPIs have been shown to be a safe and effective treatment option in the majority of patients, reflux inhibition could potentially be of value as an alternative monotherapy in some patients.
The development of better treatment alternatives for patients with GERD symptoms who have a partial response to PPI therapy would be aided by a better understanding of the pathophysiology of the condition (including the mechanism of symptom generation) and better identification of the proportion of these patients whose symptoms are functional or non-reflux related. It remains to be determined whether a more highly selected patient population would have a larger proportion of responders to reflux inhibition therapy than the broad population of patients who have a partial response to PPI therapy tested here. It is also possible that reflux inhibition may prove to be more effective when used alongside agents that target GERD symptoms by other mechanisms, for example, by modulating oesophageal sensitivity. However, based on the results of the current trial, we conclude that in patients with GERD symptoms partially responsive to PPI therapy, lesogaberan seems only marginally superior to placebo in achieving an improvement in symptoms.
Acknowledgments
This clinical trial was sponsored by AstraZeneca. The authors thank Dr Stephen Sweet of Oxford PharmaGenesis who provided writing support funded by AstraZeneca.
References
Footnotes
-
Funding This study was supported by AstraZeneca R&D, Mölndal, Sweden.
-
Competing interests Nicholas Shaheen has received grant/research support from AstraZeneca, BÂRRX Medical, Procter & Gamble Pharmaceuticals, Oncoscope, Inc., Takeda Pharmaceutical Company and CSA Medical, Inc., and has received consulting fees from Takeda Pharmaceutical Company, CSA Medical, Inc., AstraZeneca Pharmaceuticals LP, Oncoscope, Inc., NeoGenomics and Shire Pharmaceuticals Inc. Hans Denison, Karin Björck and Maria Karlsson are employees of AstraZeneca R&D, Mölndal, Sweden. Debra Silberg was an employee of AstraZeneca R&D, Wilmington, DE, USA, at the time the study was carried out.
-
Patient consent All patients provided informed consent before the initiation of study-specific procedures.
-
Ethics approval The study was carried out in accordance with the Declaration of Helsinki. A three-member Data Safety and Monitoring Board with no affiliation to the sponsor or other involvement in the trial assessed interim safety data.
-
Provenance and peer review Not commissioned; externally peer reviewed.