Article Text

Abstract
Objective The aim of this study was to assess the efficacy and safety of adalimumab (ADA), a recombinant human monoclonal antibody against tumour necrosis factor α (TNF), for the induction of clinical remission in anti-TNF naïve patients with moderately to severely active ulcerative colitis.
Methods This 8-week, multicentre, randomised, double-blind, placebo-controlled study (NCT00385736), conducted at 94 centres in North America and Europe, enrolled ambulatory adult patients with Mayo score of ≥6 points and endoscopic subscore of ≥2 points despite treatment with corticosteroids and/or immunosuppressants. Under the original study protocol, 186 patients were randomised (1:1) to subcutaneous treatment with ADA160/80 (160 mg at week 0, 80 mg at week 2, 40 mg at weeks 4 and 6) or placebo. Subsequently, at the request of European regulatory authorities, the protocol was amended to include a second induction group (ADA80/40: 80 mg at week 0, 40 mg at weeks 2, 4 and 6). The primary efficacy endpoint was clinical remission (Mayo score ≤2 with no individual subscore >1) at week 8, assessed in 390 patients randomised (1:1:1) to ADA160/80, ADA80/40, or placebo. Safety was assessed in all enrolled patients. Patients, study site personnel, investigators, and the sponsor were blinded to treatment assignment.
Results At week 8, 18.5% of patients in the ADA160/80 group (p=0.031 vs placebo) and 10.0% in the ADA80/40 group (p=0.833 vs placebo) were in remission, compared with 9.2% in the placebo group. Serious adverse events occurred in 7.6%, 3.8% and 4.0% of patients in the placebo, ADA80/40, and ADA160/80 groups, respectively. There were two malignancies in the placebo group, none in the ADA groups. There were no cases of tuberculosis and no deaths.
Conclusions ADA160/80 was safe and effective for induction of clinical remission in patients with moderately to severely active ulcerative colitis failing treatment with corticosteroids and/or immunosuppressants.
Clinical trial NCT00385736.
- Antibody targeted therapy
- clinical trials
- ulcerative colitis
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Conventional treatments for moderate to severely active ulcerative colitis have limited efficacy and are associated with adverse events.
One biological therapy, the anti-TNFα chimeric monoclonal antibody infliximab, has been proven to induce and maintain remission in ulcerative colitis patients.
Adalimumab (ADA), a fully human monoclonal antibody against TNFα, is effective in inducing and maintaining remission in moderately to severely active Crohn's disease, but its efficacy in ulcerative colitis is unknown.
What are the new findings?
This trial demonstrated that ADA is effective in inducing remission in moderately to severely active ulcerative colitis.
At week 8, 18.5% of patients in the ADA160/80 group (p=0.031 vs placebo) and 10.0% in the ADA80/40 group (p=0.833 vs placebo) were in remission, compared with 9.2% in the placebo group.
Adalimumab was well tolerated, with a safety profile comparable to those seen in clinical trials.
How might it impact on clinical practice in the foreseeable future?
Follow-up of the patients in this trial is ongoing.
Introduction
Ulcerative colitis is an idiopathic, chronic inflammatory disease of the large intestine, usually involving the rectum, characterised by a continuous pattern of inflammation and ulceration of the intestinal mucosa and submucosa. Ulcerative colitis has a significant negative impact on patient quality of life1 and places a substantial financial burden on healthcare systems, with direct cost estimates exceeding $3.4 billion in the USA and €5.4 billion in Europe.2 The goal of therapy in ulcerative colitis is to induce and maintain remission. Conventional medical therapies include 5-aminosalicylic acid, corticosteroids and oral immunosuppressants (azathioprine, 6-mercaptopurine and cyclosporine). However, these agents inadequately control the disease in a substantial proportion of patients and can lead to adverse events (AEs). Thus, there is a need for new therapies beyond conventional treatment options for many patients with ulcerative colitis.
Biological therapies targeting specific immune pathways have been tested in patients with ulcerative colitis3–5 in an attempt to increase treatment options and improve clinical outcomes, but only the chimeric monoclonal anti-tumour necrosis factor α (TNFα) antibody, infliximab, has been shown to induce and maintain remission in patients with moderate to severe ulcerative colitis.6 7 Infliximab is currently the only biological agent approved to treat patients with ulcerative colitis. TNFα is a naturally occurring proinflammatory cytokine that appears to play a critical role in the inflammatory processes of ulcerative colitis.8 TNFα expression and secretion is increased in mucosal macrophages isolated from inflammatory bowel disease lesions, and it is found in increased concentrations in the blood, mucosal tissue, and stools of patients with ulcerative colitis.9–11
Adalimumab is a fully human recombinant monoclonal antibody against TNFα. Its specific and high-affinity binding to the soluble and transmembrane forms of TNFα inhibits the ability of TNFα to bind to its receptors. Adalimumab is approved to treat rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, psoriasis, and Crohn's disease in the USA and Europe. Adalimumab has been shown to be an effective treatment for moderately to severely active Crohn's disease, with a safety profile consistent with that observed for its use in other indications.12 Several small open-label trials and case reports suggest that adalimumab can induce remission in patients with ulcerative colitis,13–15 but there are no large randomised controlled trials demonstrating its efficacy in ulcerative colitis. The objective of this study was to assess the efficacy and safety of two dosing regimens of adalimumab for the induction of clinical remission in patients with moderately to severely active ulcerative colitis.
Methods
Study design and ethics statement
This phase III, multicentre, randomised, double-blind, placebo-controlled trial (http://www.clinicaltrials.gov/) was conducted in North America and Europe, from August 2007 to February 2010. Activated centres (94) were located in the USA (34), Puerto Rico (three), Canada (five), western Europe (32), and eastern Europe (20). The study was approved by the institutional review board for each site, and carried out according to guidelines of the International Conference on Harmonisation and the ethical principles originating in the Declaration of Helsinki. All patients provided written informed consent prior to the initiation of any study-related screening procedures.
The original study protocol included one adalimumab induction group (ADA 160/80: 160 mg at week 0, 80 mg at week 2, 40 mg at weeks 4 and 6) and a placebo group. Subsequent to the initiation of the study, at the request of European regulatory authorities, the protocol was amended (Amendment 3), to include a second adalimumab induction group (ADA 80/40: 80 mg at week 0, 40 mg at weeks 2, 4 and 6) and changes to the eligibility criteria. Thus, a subset of patients was enrolled under the original protocol and another subset of patients was enrolled under the amended protocol.
Eligibility
Adult ambulatory patients with moderately to severely active ulcerative colitis, defined by a full Mayo score16 (including endoscopic assessment) of 6–12 with an endoscopy subscore of 2–3, despite concurrent and stable treatment with oral corticosteroids and/or immunomodulators, were included. Diagnosis of ulcerative colitis was confirmed by colonoscopy with biopsy or flexible sigmoidoscopy with biopsy during screening. Patients concurrently treated with oral corticosteroids were receiving a stable dose (prednisone ≥20 mg/day or equivalent for at least 14 days, or <20 mg/day for at least 40 days) prior to baseline. Patients treated with immunomodulators received at least a consecutive 90-day course (with stable dose for at least 28 days) prior to baseline of azathioprine (at least 1.5 mg/kg/day, or highest tolerated dose) or 6-MP (at least 1 mg/kg/day, or highest tolerated dose). For patients treated with both oral corticosteroids and immunomodulators, only one of the drugs was necessary for eligibility. Concurrent therapy was not required for patients who failed to respond to or could not tolerate previous corticosteroid or immunomodulator treatment, as judged by the investigator. Female patients were post-menopausal or surgically sterile, or using an approved method of birth control.
Patients with ulcerative proctitis were excluded from the study. Patients were also excluded for previous receipt of any anti-TNF agent (original protocol) or any biological agent (Amendment 3), including adalimumab; receipt of intravenous corticosteroids within 14 days prior to screening and during screening; receipt of cyclosporine, tacrolimus, mycophenolate mofetil, or methotrexate within 60 days (original protocol) or cyclosporine, tacrolimus, or mycophenolate mofetil within 30 days (Amendment 3) prior to baseline; receipt of therapeutic enema or suppository, with the exception of those required for endoscopy, within 14 days prior to screening endoscopy and during the screening period; receipt of any investigational agent within 30 days or five half lives prior to baseline. Additional exclusion criteria in both protocols were as follows: pregnancy or lactation; current receipt of total parenteral nutrition; positive Clostridium difficile stool assay; treatment of infection with intravenous (within 30 days of baseline) or oral (within 14 days prior to baseline) antibiotics, antivirals, or antifungals; history of listeria, histoplasmosis, chronic or active hepatitis B infection, HIV, immunodeficiency syndrome, central nervous system demyelinating disease, or untreated tuberculosis; history of malignancy, or signs of dysplasia or malignancy on endoscopy; or history of drug or alcohol abuse during the past year.
Randomisation and blinding
At baseline (week 0), patients were randomly assigned to receive adalimumab induction (ADA 160/80) or placebo (1:1 ratio, original protocol), or one of two adalimumab induction doses (ADA 160/80 or ADA 80/40) or placebo (1:1:1 ratio, Amendment 3), using a central randomisation scheme generated by the study sponsor. Patients, study site personnel, study investigators, and the study sponsor were blinded to treatment assignment throughout the study.
Study drug and placebo were provided as subcutaneous (SC) injection solution in pre-filled syringes, containing either adalimumab (40 mg/0.8 ml) or placebo (figure 1). Patients enrolled in the ADA 160/80 group received adalimumab (160 mg SC total, in one day) at week 0, followed by adalimumab (80 mg SC total, in one day) at week 2, and adalimumab (40 mg SC total, in one day) at weeks 4 and 6. Patients in the ADA 80/40 group received adalimumab (80 mg SC total, in one day) and placebo at week 0, adalimumab (40 mg SC total, in one day) and placebo at week 2, and adalimumab (40 mg SC total, in 1 day) at weeks 4 and 6. To maintain blinding under both protocols, patients in the placebo group received the same number of injections as patients in the adalimumab treatment group(s). Dosage(s) of concomitant medication(s) for ulcerative colitis remained unchanged throughout study.

Study design through week 8, original protocol and after Amendment 3. Patients continued in an extension phase (open-label adalimumab, 40 mg every other week) through week 52, beginning at week 12 (original protocol) or week 8 (Amendment 3).
Study procedures
Endoscopy was performed at screening and week 8. At screening, colonoscopy was performed in patients without a colonoscopy report available within 6 months of screening, and flexible sigmoidoscopy was performed in all others. Mayo scores16 were recorded at weeks 0 and 8. At each clinic visit (baseline (week 0) and weeks 2, 4, 6 and 8), patients underwent physical examination; vital signs, previous and concomitant medications, partial Mayo scores (Mayo score without the endoscopy subscore), and AEs were recorded. At weeks 0, 4 and 8, general laboratory tests and urinalysis were performed, and C-reactive protein (CRP; high sensitivity) was measured. Efficacy endpoints were assessed at week 8. Patients in the study continued in an open-label phase on adalimumab 40 mg every other week through week 52, beginning at week 12 (original protocol) or week 8 (Amendment 3).
Study populations
Because Amendment 3 added a second adalimumab dose group and included changes to the eligibility criteria, patients enrolled before the amendment were not included in the primary analysis data set. Thus, the intention-to-treat (ITT-A3) population for the primary efficacy evaluations included patients with confirmed ulcerative colitis enrolled under Amendment 3 or later who received at least one injection in the ADA 160/80, ADA 80/40, and placebo groups. All patients with confirmed ulcerative colitis randomly assigned to a study group under the original protocol and all its amendments who received at least one dose of study drug or placebo were included in the second intention-to-treat population, ITT-E, so that patients enrolled under the original protocol could also be assessed. The safety population included all patients under the original protocol and all its amendments who received at least one dose of study drug or placebo.
Efficacy variables
The primary efficacy variable was the proportion of patients in each treatment group in remission per Mayo score at week 8 (remission defined as Mayo score ≤2 with no individual subscore >1), assessed in the ITT-A3. The primary efficacy variable was assessed in the ITT-E population as a sensitivity analysis. Ranked secondary variables were assessed at week 8 in each group of the ITT-A3 and included: proportion of patients with clinical response per Mayo score (response: decrease in Mayo Score ≥3 points and ≥30% from baseline PLUS a decrease in the rectal bleeding subscore ≥1 or an absolute rectal bleeding subscore of 0 or 1); proportion of patients with mucosal healing (endoscopy subscore of 0 or 1); proportion of patients with subscores indicative of mild disease (rectal bleeding subscore ≤1, physician's global assessment (PGA) subscore ≤1, or stool frequency subscore ≤1).
Additional analyses
All additional analyses were performed for the ITT-A3 population. The proportion of patients in remission per partial Mayo score (defined as partial Mayo score ≤2 with no subscore >1) at weeks 0, 2, 4, 6 and 8 was determined in all groups. The proportion of patients in remission per Mayo score at week 8 was assessed after stratification by baseline Mayo scores (6–9 vs 10–12), by baseline concomitant medication (corticosteroid and/or immunomodulators, and aminosalicylates), by the extent of disease at baseline, by baseline CRP level (<10 mg/l vs ≥10 mg/l), and by baseline weight (tertiles). The proportion of patients achieving the secondary efficacy endpoints at week 8 was assessed in the placebo and ADA 160/80 groups in four regions (Canada, eastern Europe, USA/Puerto Rico, western Europe). The median change from baseline in CRP was assessed at week 8.
Sample size
For the primary endpoint, sample size calculation in Amendment 3 assumed 15% of patients in the placebo group would achieve clinical remission at week 8. A sample size of 125 in each treatment group was adequate to detect a 15% difference using a χ2 test with 80% power at a 0.05 two-sided significance level.
Statistical methods
Demographics and baseline characteristics (ITT-A3 and ITT-E) were summarised using descriptive statistics. Continuous variables were compared using ANOVA, discrete variables using the χ2 test. Efficacy variables (primary and secondary) and partial Mayo scores were assessed in the ITT-A3 population. Results for the ADA 160/80 and ADA 80/40 groups were compared with results for the placebo group using the χ2 test for dichotomous endpoints, with missing or incomplete data handled using non-responder imputation; remission rates in the ITT-E population were analysed using the same methods. In the subgroup analyses (baseline Mayo scores, extent of disease at baseline, baseline concomitant medications, baseline CRP level, and baseline weight), 95% confidence intervals for the difference in proportions between active treatment groups and placebo were based on normal approximation to the binomial distribution. Treatment difference between placebo and ADA 160/80 groups in secondary efficacy variables in the ITT-A3 population within four geographical regions (Canada, eastern Europe, USA/Puerto Rico, western Europe), were compared using the same methods as for the primary efficacy variable. Median changes in CRP were calculated using both as-observed analysis and last observation carried forward; results in the adalimumab treatment groups were compared with placebo results using the Kruskal–Wallis test.
The number and percentage of patients experiencing treatment-emergent AEs was determined in the safety population. Incidence of AEs in the placebo group was compared with the incidence of AEs in each adalimumab dose group using Fisher's exact test.
Results
Patient flow
Of 576 patients enrolled in the study, 90% (521) completed 8 weeks (figure 2). The most common reasons for discontinuation were AEs and lack of efficacy. The ITT-A3 population (N=390) and the ITT-E population (N=575) included patients with confirmed ulcerative colitis enrolled under the original protocol and its amendments who received at least one dose of study drug. The safety population included all patients who received at least one dose of the study drug (N=576); one patient who received study drug but had Crohn's disease, not ulcerative colitis, was included in the safety population but not the ITT populations. The blind was broken for three patients, one from each group, because of AEs; these patients did not complete the study, but were included in the ITT and safety populations.
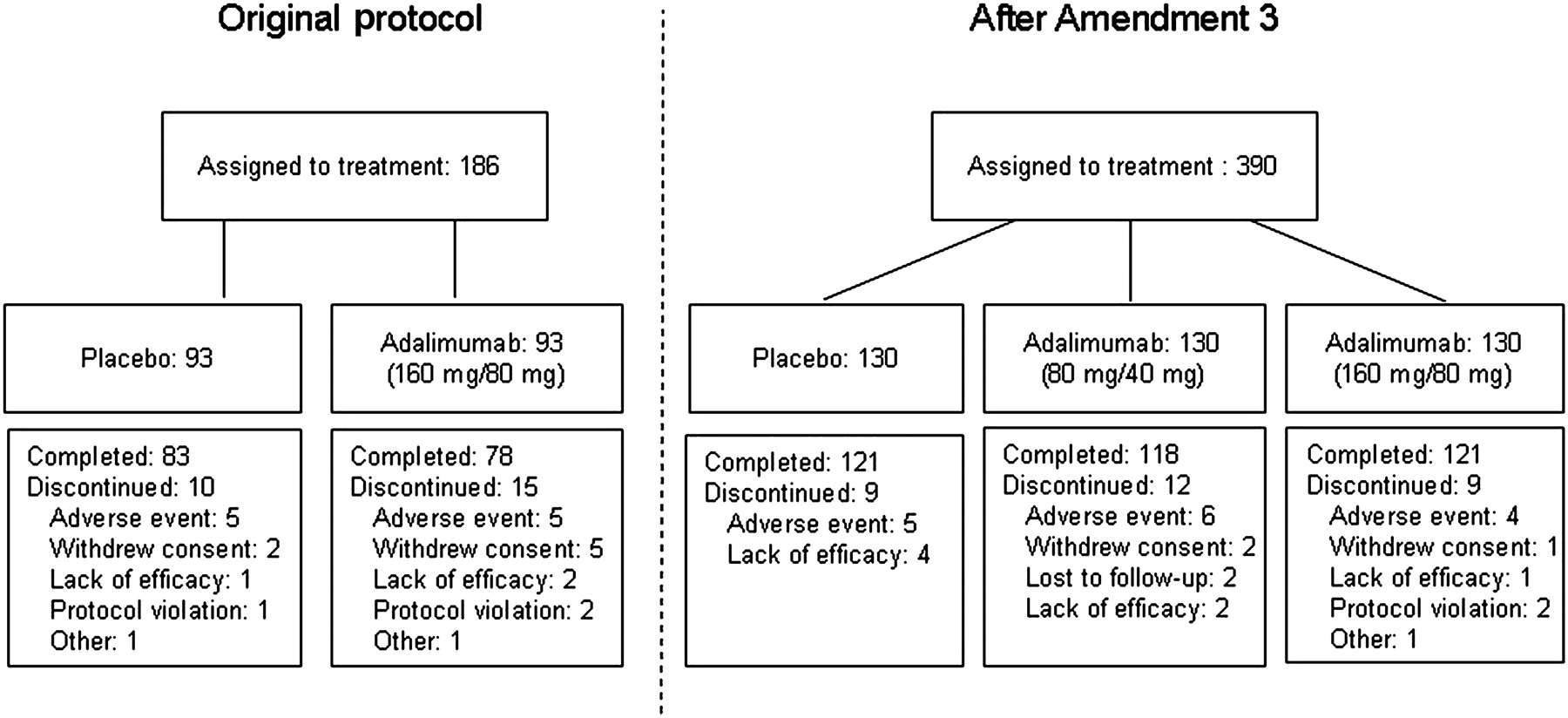
Patient disposition before and after protocol Amendment 3.
Baseline characteristics
The baseline demographic and clinical characteristics of patients in the ITT-A3 population were similar across the three treatment groups (table 1). Patients in the ADA 80/40 group had a numerically longer duration of disease, a higher mean Mayo score, and a higher median CRP concentration at baseline, but the differences were not statistically significant. Baseline demographics in the ITT-E and safety populations were similar to those of the ITT-A3 population (supplementary material).
Baseline demographics and clinical characteristics of patients in the ITT-A3 population
Outcomes
Approximately twice as many patients in the ADA 160/80 group achieved clinical remission at week 8 (primary endpoint) compared with patients in the placebo group (p=0.031; figure 3). The proportion of patients in clinical remission at week 8 in the placebo and ADA 80/40 groups was similar (p=0.833). Clinical remission in the ITT-E population was generally similar to that of the ITT-A3 population for each treatment group (supplementary material).
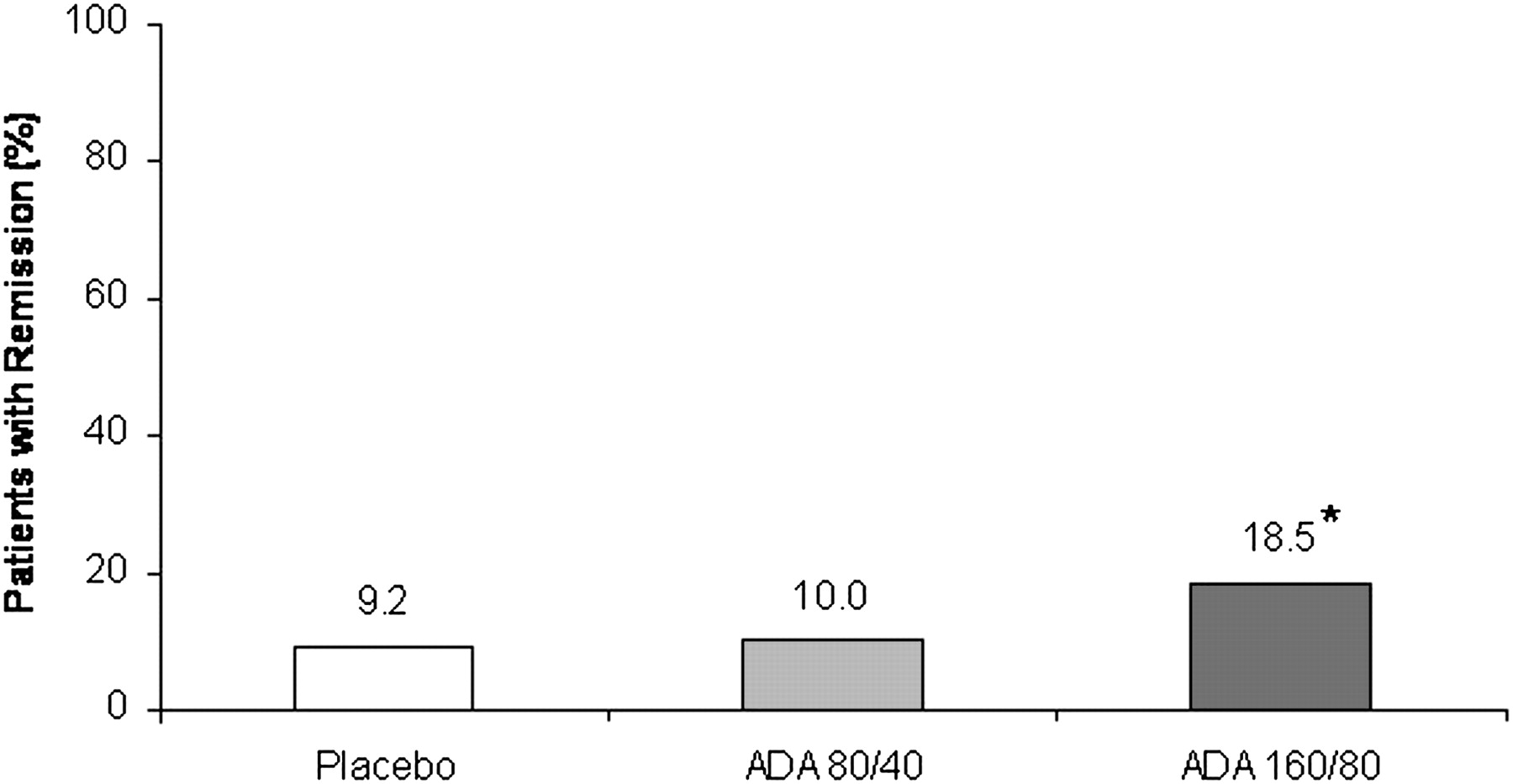
Clinical remission at week 8 in the ITT-A3 population (non-responder imputation). N=130 for each group. *p=0.031 versus placebo.
A higher percentage of patients in the ADA 160/80 group achieved clinical response and subscores indicative of mild disease (≤1) for all components of the Mayo score, compared with the placebo group, but the percentage was significantly different only for the Mayo rectal bleeding subscore (p=0.038) and PGA subscore (p=0.035, table 2).
Summary of secondary efficacy results
Additional analyses
Partial Mayo scores were used to gauge induction of remission over time. The proportion of patients in remission (partial Mayo score ≤2 with no subscore >1) increased over time in both adalimumab groups, with statistically significant separation between the ADA 160/80 and placebo groups from week 2 through week 8 (figure 4). The proportion of patients in remission in the placebo group reached a maximum at week 6, and then declined between weeks 6 and 8.

Clinical remission per partial Mayo score (≤2 with no subscore >1) over time in the ITT-A3 population (non-responder imputation). N=130 for each group. *p<0.05; **p<0.01 versus placebo.
Patients with Mayo score ≥10 at baseline had lower rates of remission compared with patients with less active disease in all groups (table 3), though this was most pronounced in the ADA 80/40 and the placebo groups. Treatment effect was more pronounced in patients without extensive colitis, in patients treated with immunomodulator (IMM) without corticosteroids at baseline, and in those who did not receive aminosalicylates at baseline. High CRP (≥10 mg/l) at baseline and higher baseline weight (≥82.0 kg) were associated with reduced remission rates, especially in the ADA 160/80 group.
Subgroup analysis results: Remission at week 8, stratified by baseline Mayo score, extensive colitis, concomitant medications, baseline CRP level, and weight tertiles
The median changes at week 8 in CRP (high sensitivity) from baseline in the ITT-A3 population, using last observation carried forward analysis, were as follows: placebo, −0.09 mg/l (range: −274.79 to 88.71); ADA 80/40, −0.49 mg/l (range: −115.76 to 88.03); and ADA 160/80, −0.77 mg/l (range: −95.09 to 130.41; p=0.018 for ADA 160/80 vs placebo). The results using as observed analysis were similar.
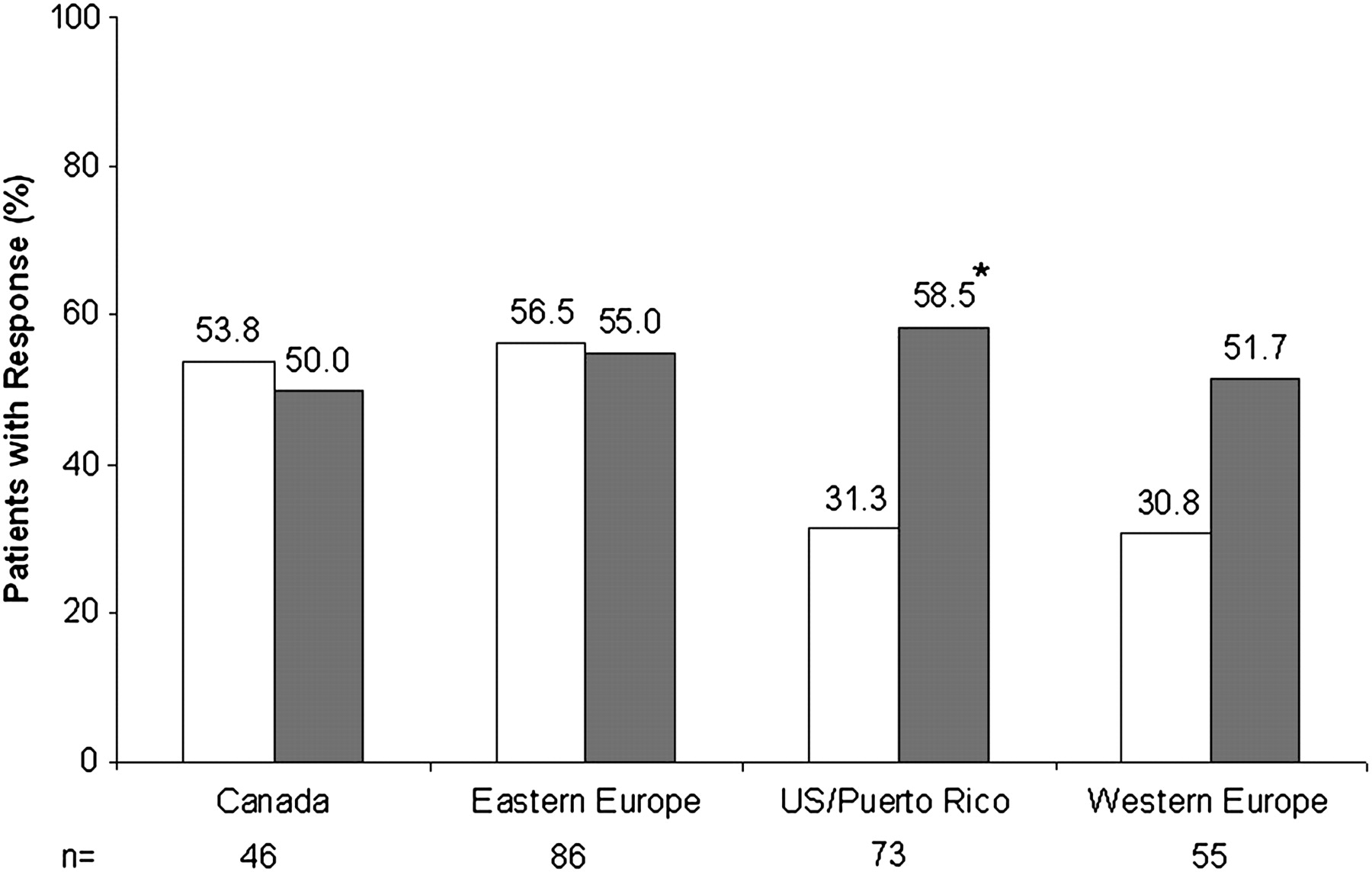
Although clinical response rates in the ADA 160/80 mg group were similar in subgroups of patients from study sites in different geographical regions (figure 5), placebo response rates in eastern Europe and Canada were high compared with the other regions. The same pattern was observed in other secondary endpoints assessed by region: the proportion of placebo patients with mucosal healing, rectal bleeding subscore ≤1, or PGA subscore ≤1 at week 8 was notably high in both eastern Europe and Canada, compared with the other regions (data not shown). Patient allocation across the three treatment groups in each region was similar.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical response at week 8, stratified by region. White bars, placebo; grey bars, ADA 160/80. *p<0.05 versus placebo. (Eastern Europe: Czech Republic, Hungary, Slovakia, Poland. Western Europe: Austria, Belgium, Germany, Italy, Netherlands, Sweden.)
Safety
Adalimumab treatment was generally well tolerated at both induction doses and the overall safety profile of adalimumab was comparable to that of placebo. Analyses of laboratory parameters and vital signs did not reveal any safety issues. A similar proportion of patients in each study group experienced treatment emergent AEs (table 4). The proportion of patients who discontinued the study because of AEs was low and similar across the study groups. Ulcerative colitis was the most common AE leading to discontinuation in 4.0% of placebo, 3.8% of ADA 80/40, and 3.6% of ADA 160/80 patients. The incidence of injection site pain was low and similar across the three study groups.
Summary of treatment-emergent adverse events (safety population: all patients who received at least one dose of study drug or placebo)
The incidence of serious AEs was highest in the placebo group and almost double the incidence of serious AEs observed in the ADA 160/80 group, but the differences were not statistically significant. Serious infections were reported in three patients in the placebo group, two patients in the ADA 80/40 group, and none in the ADA 160/80 group. Malignancies occurred in two placebo-treated patients, with none in the adalimumab groups. One patient in the ADA 160/80 group experienced an opportunistic infection (oesophageal candidiasis). There were no cases of tuberculosis in the study, and no deaths.
Discussion
In this study, adalimumab (160/80 mg) was effective for the induction of remission in patients with moderately to severely active ulcerative colitis who were not responding to, or had previously failed to respond to, or were intolerant to conventional therapies. Both induction doses of adalimumab used in this short-term induction study were well tolerated. The incidence rates of all infections and serious infections were comparable between the adalimumab treatment groups and the placebo group.
In contrast with the infliximab studies, which demonstrated that for induction of remission of ulcerative colitis, 10 mg/kg of infliximab do not provide higher efficacy than 5 mg/kg,7 17 dosing higher than 160/80 mg of adalimumab has not been tested. Dosing in the adalimumab induction trial in patients with ulcerative colitis was based on the doses of adalimumab known to be safe and effective in Crohn's disease.18–20 By the fourth week of adalimumab induction, up to one third of patients with Crohn's disease achieve remission, and approximately one half achieve a clinical response.19 20 In patients with Crohn's disease, only the 160/80 mg dose of adalimumab demonstrated efficacy for the induction of remission,19 although improvement in clinical response (defined as a decrease of ≥70 points in the Crohn's disease acitivity index score) was observed as early as week 1 with the 80/40 mg dose. In contrast, there was little or no separation from placebo in any of the primary and secondary outcomes for the 80/40 mg dose in the ulcerative colitis trial. Furthermore, in our analysis by baseline weight tertiles, we found rates of clinical remission for patients below 82 kg to be more than twice the rate for patients above 82 kg in the 160/80 mg dose group (table 3). In addition, the results from the CRP subgroups suggest that patients with elevated CRP may have an inflammatory disease burden that is not adequately addressed by the doses used in this study. Thus, our data suggest the possibility that a substantial proportion of patients with ulcerative colitis may require a higher dose of adalimumab to induce remission, compared with Crohn's disease patients, though the biological rationale for this remains unclear. A 52-week, randomised, double-blind trial to assess the ability of adalimumab to induce and maintain remission in patients with ulcerative colitis, including those previously exposed to infliximab, is in progress; pharmacokinetic data from that trial may help clarify currently unanswered questions about dosing of adalimumab for ulcerative colitis.
In addition to increased dose, patients with ulcerative colitis may require a longer exposure to high doses of adalimumab than patients with Crohn's disease to achieve induction of remission. The partial Mayo score data in this study indicate that the plateau of efficacy of adalimumab had not yet been reached by week 8 (figure 4), suggesting a need for longer exposure to adalimumab to observe a maximum response. Patients in this 8-week induction trial continued in a 52-week open-label maintenance phase, so follow-up is ongoing. Results from the open-label phase of the induction trial, and the double-blind maintenance trial currently in progress, are expected to improve our understanding of the peak period for induction of remission in patients with moderately to severely active ulcerative colitis.
While secondary endpoint measures in this study were consistently higher in the ADA 160/80 group compared with the ADA 80/40 or placebo groups, a statistically significant difference was observed only for the rectal bleeding subscore and the PGA (table 2). In the placebo groups in the infliximab ACT 1 and ACT 2 trials,7 the clinical remission rates at week 8 (14.9% and 5.7%, respectively) were in line with the 9.2% we observed in our placebo group. By contrast, the proportion of placebo patients achieving clinical response at week 8 in ACT 1 and ACT 2 (primary endpoint) were lower than our observed results (37.2% and 29.3%, respectively, vs 44.6% in our results). Likewise, the proportion of patients achieving mucosal healing in the placebo groups at week 8 was 33.9% in ACT 1 and 30.9% in ACT 2, compared with 41.5% in our study. The high placebo rates we observed in the secondary endpoints most likely contributed to the lack of statistical significance.
A recent meta-analysis of placebo response rates in randomised clinical trials in ulcerative colitis21 reported a pooled placebo remission rate of 23% and a pooled placebo improvement rate of 32% (‘remission’ and ‘improvement’ definitions varied between the studies included in the analysis, but were generally based on disease activity indices or scores). Higher placebo remission and improvement rates were associated with trials carried out exclusively in Europe compared with studies performed exclusively in the USA, while endoscopic remission in placebo patients was greater in trials of four or more weeks. The remission rate in our placebo patients was lower than the 23% pooled placebo remission rate, while all of our secondary endpoints had placebo response rates higher than the 32% pooled improvement rate reported by Garud and colleagues.21 In our study, while clinical response rates of the ADA 160/80 treatment arm were similar across the four regions analysed (50% to 58.5%), the highest clinical response rates for the placebo patients were observed in eastern Europe and Canada (58.7% and 53.8%, respectively, vs 26.9% in western Europe and 31.3% in USA/Puerto Rico) (figure 5). The reasons for different placebo response rates in different geographical regions are unclear.
Garud et al21 did not find a correlation between number of study visits and placebo response rates, but an earlier meta-analysis of the placebo response in ulcerative colitis22 reported increased placebo response in endoscopic remission, histological remission, and clinical benefit (defined as response, improvement or clinical remission), in studies with more than three visits, compared with studies with three or fewer visits. It is possible that a high number of clinic visits (five visits in 8 weeks) in our trial contributed to a placebo effect via the psychological and neurobiological mechanisms thought to influence responses in placebo-treated patients.23 Similarly, the placebo effect might have been enhanced by high expectations for success among investigators and patients in the adalimumab trial, based upon the knowledge of the efficacy of infliximab in ulcerative colitis.
In summary, this trial demonstrated that adalimumab (160 mg/80 mg) is effective for induction of remission in patients with moderately to severely active ulcerative colitis failing concurrent treatment with or intolerant to oral corticosteroids and/or immunomodulators. However, the unusually high response rates in the placebo group did not allow robust conclusions to be drawn for the secondary endpoints in the trial. Nevertheless, adalimumab was well tolerated, with a safety profile comparable to those seen in clinical trials and clinical experience with adalimumab in Crohn's disease and other indications.12
Acknowledgments
The authors would like to thank Wenrui Wang, of Abbott, for statistical analysis and programming support, and Laurinda Cooker, of Abbott, for assistance in the writing and preparation of this manuscript. Ashish Kumar provided statistical analysis and programming support and was compensated by Abbott for his work.
References
Supplementary materials
online only appendix
Files in this Data Supplement:
Footnotes
See Commentary, p 741
Linked articles 233403.
Funding This study was funded by Abbott.
Competing interests The authors declare that WR, WJS, DH, GD, SH, SS, RP and RNF have received support from Abbott for the submitted work; and MBT, BH, WK, AL and RT are employees of Abbott.
Ethics approval This study was conducted with the approval of the institutional review board for each participating site, and carried out according to guidelines of the International Conference on Harmonization and the ethical principles originating in the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.