Article Text

Abstract
Background: Exposure to elevated levels of ambient air pollutants can lead to adverse cardiovascular effects. Potential mechanisms include systemic inflammation and perturbation of the coagulation balance.
Objectives: To investigate long- and short-term effects of air pollution exposure on serum levels of inflammatory (IL-6, TNF-α and CRP) and coagulation (fibrinogen and PAI-1) markers relevant for cardiovascular pathology.
Methods: The study group consisted of a population sample of 1028 men and 508 women aged 45–70 years from Stockholm. Long-term air pollution exposure was assessed using spatial modelling of traffic-related NO2 and heating-related SO2 emissions at each subject’s residential addresses over retrospective periods of 1, 5 and 30 years. Short-term exposure was assessed as averages of rooftop measurements over 12–120 h before blood sampling.
Results: Long-term exposures to both traffic-NO2 and heating-SO2 emissions showed consistent associations with IL-6 levels. 30-year average traffic-NO2 exposure was associated with a 64.5% (95% CI 6.7% to 153.8%) increase in serum IL-6 per 28.8 μg/m3 (corresponding to the difference between the 5th and 95th percentile exposure value), and 30-year exposure to heating-SO2 with a 67.6% (95% CI 7.1% to 162.2%) increase per 39.4 μg/m3 (5th–95th percentile value difference). The association appeared stronger in non-smokers, physically active people and hypertensive subjects. We observed positive non-significant associations of inflammatory markers with NO2 and PM10 during 24 h before blood sampling. Short-term exposure to O3 was associated with increased, and SO2 with decreased, fibrinogen levels.
Conclusions: Our results suggest that exposure to moderate levels of air pollution may influence serum levels of inflammatory markers.
Statistics from Altmetric.com
Numerous studies have demonstrated an association between day-to-day variations in air pollution concentrations and respiratory as well as cardiovascular morbidity and mortality.1 2 Long-term exposure to air pollution has been related to cardiovascular and cardiopulmonary mortality, reduced lung function development and symptoms of bronchitis.2 3 4 5 However, there is limited evidence on possible mechanisms for the effect of long-term exposure. One epidemiological study assessing carotid intima-media thickness showed an association between ambient air pollution and atherosclerosis in humans.6
What this paper adds
Epidemiological studies have demonstrated an association between air pollution exposure and cardiovascular morbidity and mortality, but there is limited evidence on the possible mechanisms of such effects.
Inflammatory and coagulation processes are known to be of relevance for cardiovascular pathology, so this study aimed to investigate the association of long- and short-term air pollution exposure with blood markers of inflammation and coagulation.
Long-term exposure to local traffic- or residential heating-related air pollution appeared to increase serum interleukin (IL)-6 levels, and a suggested positive association was seen also for short-term exposure to NO2 and PM10 with IL-6, tumour necrosis factor-α and C-reactive protein levels.
The results imply that the disturbed cytokine milieu caused by air pollution exposure may have negative consequences for cardiovascular health both in the long and short term.
The reported association of elevated inflammatory cytokine IL-6 levels with long-term exposures to air pollutants in Stockholm, a European city with relatively clean air, provides further support for the importance of limiting urban air pollution levels to protect health.
On a functional level, adverse short- and long-term exposure effects of air pollution may be linked to several potential mechanisms. For example, air pollution induces pulmonary inflammation leading to a systemic cytokine-mediated inflammatory response,1 3 which can contribute to atherosclerosis.7 8 Another proposed mechanism involves effects on cardiac autonomic control and increased risk of arrhythmia, either as a result of airway receptor stimulation or as a consequence of the inflammatory response.1 Investigating changes in blood markers, including inflammation and coagulation parameters, may help to elucidate the pathways of disease development caused by air pollution exposure.1 3 Such effects have been addressed in many epidemiological and experimental studies for short-term exposure to air pollution, but there is a lack of studies investigating long-term effects.
The present study aimed to investigate how long-term (years) and short-term (hours to days) air pollution exposure may affect the levels of some blood markers of interest for cardiovascular disease, that is, the proinflammatory cytokines interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α), C-reactive protein (CRP) and the blood coagulation factors fibrinogen and plasminogen activator inhibitor-1 (PAI-1).
Methods
Study population
The study group consisted of all subjects with available data on the blood markers of interest from the control group of the Stockholm Heart Epidemiology Program (SHEEP), a large population-based case–control study described in detail elsewhere.9 Briefly, SHEEP included all first-time myocardial infarction cases aged 45–70 years identified in Stockholm County during 1992–1994. Controls were selected randomly from the study base (Stockholm County population, 1992–1994), matched on sex, age (±5 years) and hospital catchment area. Participation rates were relatively high, with information from questionnaires and medical examinations available for 68% and 64% of male and female controls, respectively.9 The study was approved by the Ethical Committee at Karolinska Institutet and all subjects gave informed consent.
Data collection and exposure assessment
Data on lifestyle factors including heredity, diet, physical inactivity, job strain, smoking status, occupational exposures and other cardiovascular risk factors were collected from questionnaires completed by the participants. Blood samples were collected during medical examinations at outpatient clinics, which also included blood pressure measurements and anthropometric tests, for example weight and height used to calculate body mass index (BMI). A family history of coronary heart disease was defined as having one or more first-degree relatives affected by coronary heart disease before the age of 65. Physical inactivity was defined as reported inactive leisure time, including at most occasional walks, during the last 5–10 years. Diabetes mellitus was identified either from questionnaire information on controlling diabetes with insulin or other drug treatment or diet, or was newly discovered at examination (reported insulin or drug treatment, or high fasting blood glucose). Job strain was assessed with the Swedish version of the Karasek-Theorell questionnaire,10 and considered present if a subject scored above 0.765 (corresponding to the 75th percentile among all controls).9 Current smoking was defined as reported smoking within the last year. Subjects who had stopped smoking more than 1 year before inclusion were characterised as ex-smokers. Subjects receiving antihypertensive treatment according to questionnaire information or with systolic blood pressure ⩾140 mm Hg or diastolic blood pressure ⩾90 mm Hg measured at examination were classified as hypertensive. Blood analyses were also performed to determine the levels of low density lipoprotein (LDL) cholesterol, high density lipoprotein (HDL) cholesterol, triglycerides and insulin.
Long-term exposure to source-specific air pollution was assessed at the participants’ historical home addresses using a geographical information system. The detailed methods of assessment have been described previously.11 12 Briefly, source-specific emission databases were established for each decade since 1960, providing information for dispersion modelling of annual mean levels of locally emitted air pollutants.11 All addresses inhabited by study participants for more than 2 years were transformed into geographical coordinates and calibrated dispersion models for air pollution were used to estimate the annual mean level at each address.12 We calculated average exposures for the preceding 1, 5 and 30 years before study inclusion. At most 5 of 30 years of address information was allowed to be missing.12 For the 5-year means, at most 1 year of address information was allowed to be missing. The missing air pollution data that resulted for these years of unknown residency were replaced by the mean among the controls for that calendar year.12 For the 1-year mean, no missing address information was allowed. In this study, we analysed two air pollutants characterising different exposure sources: NO2 from local traffic emissions (traffic-NO2) as a marker of the mixture of vehicle emissions, and SO2 from local residential combustion heating (heating-SO2) as a marker mainly reflecting oil combustion.
Short-term air pollution exposure measures were based on routine hourly rooftop measurements of total ambient NO2, PM10, O3 and SO2 in Stockholm. The monitors are located on two sites selected to be representative of the urban background in the centre of Stockholm. PM10 measurements started in the spring of 1994 and PM10 exposure can thus only be calculated for subjects included in the study after that point in time. Average exposures were calculated for intervals of 0–12 h and 12–24 h before the hour of blood sampling, as well as for the previous 48 h (2 days) and 120 h (5 days). Since the blood sampling was carried out in the morning, the preceding 0–12 h interval corresponds to evening, night and morning exposures, and the 12–24 h interval corresponds to the day-time exposure average. Subjects with 75% or more of hourly measurements available within a particular averaging interval were included and the mean was calculated from the available values.
Assessment of outcome blood markers
Serum IL-6 was measured using an enzyme-linked immunosorbent assay (IL-6 Eli-pair, Diaclone Research, Besancon, France).13 TNF-α was detected in serum with the Quantikine HS human TNF-alfa kit (R&D Systems, Minneapolis, MN).14 C-reactive protein (CRP) was measured on EDTA plasma samples using a high sensitivity immunonephelometric assay (Dade-Behring, Marburg, Germany) on the automated BN II system from Dade-Behring.13 An assay of fibrinogen fibrin polymerisation time (FPT test) according to the method described by Vermylen et al15 was used to measure plasma fibrinogen levels,16 and the Spectrolyze PAI-1 kit (Biopool AB, Umea, Sweden) was used for determining PAI-1 activity in citrated plasma samples.17
Statistical analysis
We analysed the association between long- or short-term air pollution exposure and levels of blood markers of inflammation (IL-6, TNF-α and CRP) and coagulation (fibrinogen and PAI-1). Linear regression models were used to determine effect estimates for risk factors on the levels of blood markers, which were log-transformed to achieve a closer to normal distribution. Estimated effects for both long-term source-specific as well as short-term ambient air pollution exposures were calculated as the change in the log level of an outcome blood parameter (presented as per cent change), per exposure increment in each respective air pollution exposure corresponding to the difference between the 5th and the 95th percentile exposure value, to allow for some comparability across the different exposure metrics. Since a relatively large number of factors have been reported to affect our studied markers in previous research, we initially constructed predictive models for IL-6, TNF-α, CRP, fibrinogen and PAI-1 based on a wide range of lifestyle and clinical parameters available in the study. Variables contributing in a relevant way in explaining the change in blood marker levels were included in the models (with relevance level set at >0.9% change in adjusted R2). Thus, the models for different blood marker outcomes contained different sets of confounding variables; age and sex were, however, included in all models.
Once the basic predictive model had been identified, we modelled air pollution exposure by adding air pollution variables for different time intervals to the models. The assessed-long term source-specific exposure intervals comprised 1, 5 and 30 years retrospectively for all of the blood markers. For the short-term air pollution analysis, we hypothesised that fluctuation of inflammatory markers IL-6 and TNF-α can be detected after shorter intervals of time and used primarily the 0–12 and 12–24 h intervals preceding blood sampling.18 We expected that longer time intervals are needed to detect changes in coagulation markers and CRP, and thus primarily hypothesised 2- and 5-day averages for air pollution exposure for these markers.18 Variables representing factors likely to influence the blood markers and possibly associated with air pollution levels were tested for confounding by the criterion of inducing a 10% change in the estimated air pollutant effect per unit of air pollution exposure (ie, a change in exposure from the 5th to the 95th percentile of population exposure) in the linear regression models (corresponding to ±0.095 β coefficient change on the log scale). However, the analysis of confounders did not identify any additional factors to be added to the set of predictors in the initial modelling.
The final effect estimates for air pollution exposure and their 95% confidence intervals were obtained in crude (unadjusted) models and in models adjusted for all identified predictive variables from the respective predictive models, as well as age and gender. All estimates are given for the 5th to 95th percentile difference according to the exposure distribution among the exposed control subjects with complete data during that specific time period. Effect modification was assessed for a number of demographic and lifestyle factors using interaction terms in a common model. To assess the potential misclassification of short-term exposure due to varying distance from central air pollution monitors between subjects, we performed an analysis restricted to the three hospital catchment areas located closest to the monitoring locations. Statistical analysis was performed with STATA 9 software (StataCorp, College Station, TX).
Results
The study group consisted of 1028 men and 508 women with mean (SD) ages of 59 (7) and 62 (7) years, respectively (table 1). A family history of coronary heart disease was present in 26% of the men and 29% of the women. More than half of the study participants had hypertension, while diabetes mellitus occurred in less than 10%. More women than men were exposed to job strain. Current smoking rates were similar in men and women, whereas alcohol consumption was higher in men. Low physical activity was present among 33% of the men and 41% of the women. Regarding the blood markers, the mean levels of IL-6 were somewhat higher in women, whereas TNF-α, CRP, fibrinogen and PAI-1 levels were similar in the two gender groups. Only CRP and fibrinogen levels were highly correlated (see online supplementary table S1), presumably since the synthesis of both is induced by IL-6 in the systemic inflammatory response.
Demographics, risk factors and laboratory measurements for a population sample in Stockholm 1992–94
The mean source-specific residential exposures to traffic-NO2 during the last 1, 5 and 30 years ranged between 12.6 and 15.2 μg/m3, and mean exposures to heating-SO2 during the last 1, 5 and 30 years were 2.8, 4.7 and 25.8 μg/m3, respectively (table 2). The generally lower levels for the long-term estimates of traffic-NO2 and heating-SO2 than for the average short-term measurements is explained by the fact that the long-term estimates only include the local contributions from traffic and heating, while the short-term measurements are based on air pollution monitor levels and consequently also include regional and long-range contributions. The heating-SO2 average was considerably higher for the 30-year average than for the shorter averages due to the higher content of sulphur in oil used for heating in the past and because more houses had individual oil-heating systems rather than being connected to large-scale district heating systems. The estimated long-term traffic-NO2 and heating-SO2 exposures were highly correlated (see online supplementary table S2). Short-term measures in general showed low correlation except between different averaging times for the same pollutant (see online supplementary table S2). There was basically no correlation between the long-term and the short-term measures.
Descriptive statistics for long- and short-term air pollution exposure of subjects in Stockholm
We found significantly higher IL-6 levels and positive effect estimates (one statistically significant) for CRP levels after long-term exposure to elevated residential levels of traffic-related NO2 as well as combustion heating-generated SO2 (table 3). The 30-year exposure to traffic-NO2 was associated with an IL-6 level increased by 64.5% (95% CI 6.7% to 153.8%) per 28.8 μg/m3 traffic-NO2 (corresponding to the difference between the 5th and the 95th percentile exposure value), and 30-year exposure to heating emissions (measured as heating-SO2) increased the level of IL-6 by 67.6% (95% CI 7.1% to 162.2%) per 39.4 μg/m3 heating-SO2 (5th to 95th percentile value difference). Both crude and adjusted effect estimates are presented in online supplementary table S3. The traffic-NO2 effects appeared to be more pronounced in non-smokers, physically active people, and in hypertensive individuals (fig 1), although the differences in effect were not statistically significant. The pattern of effect modification was similar for heating-SO2 (data not shown). Long-term exposure to source-specific air pollution was not associated with significant differences in the levels of TNF-α, fibrinogen or PAI-1.
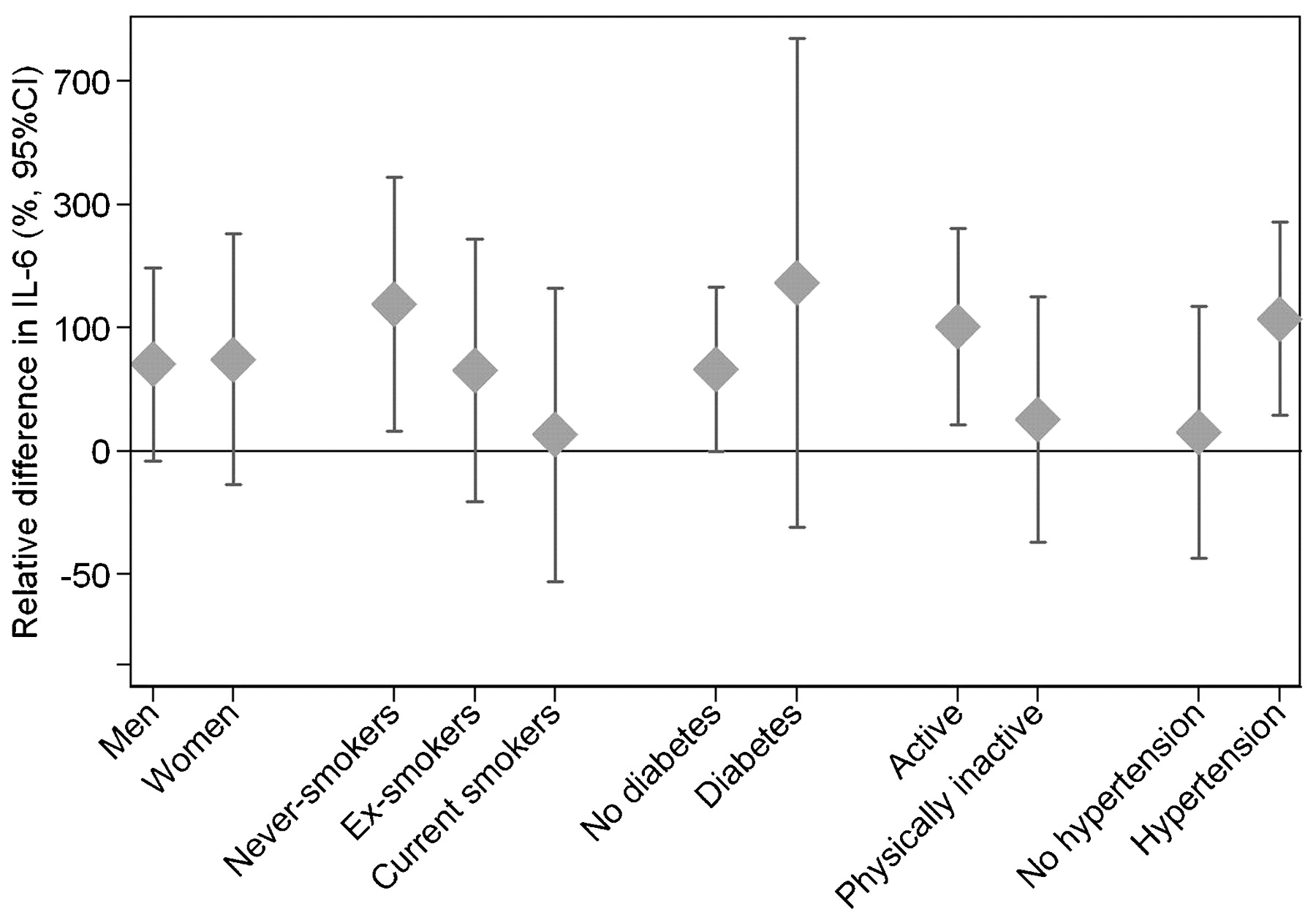
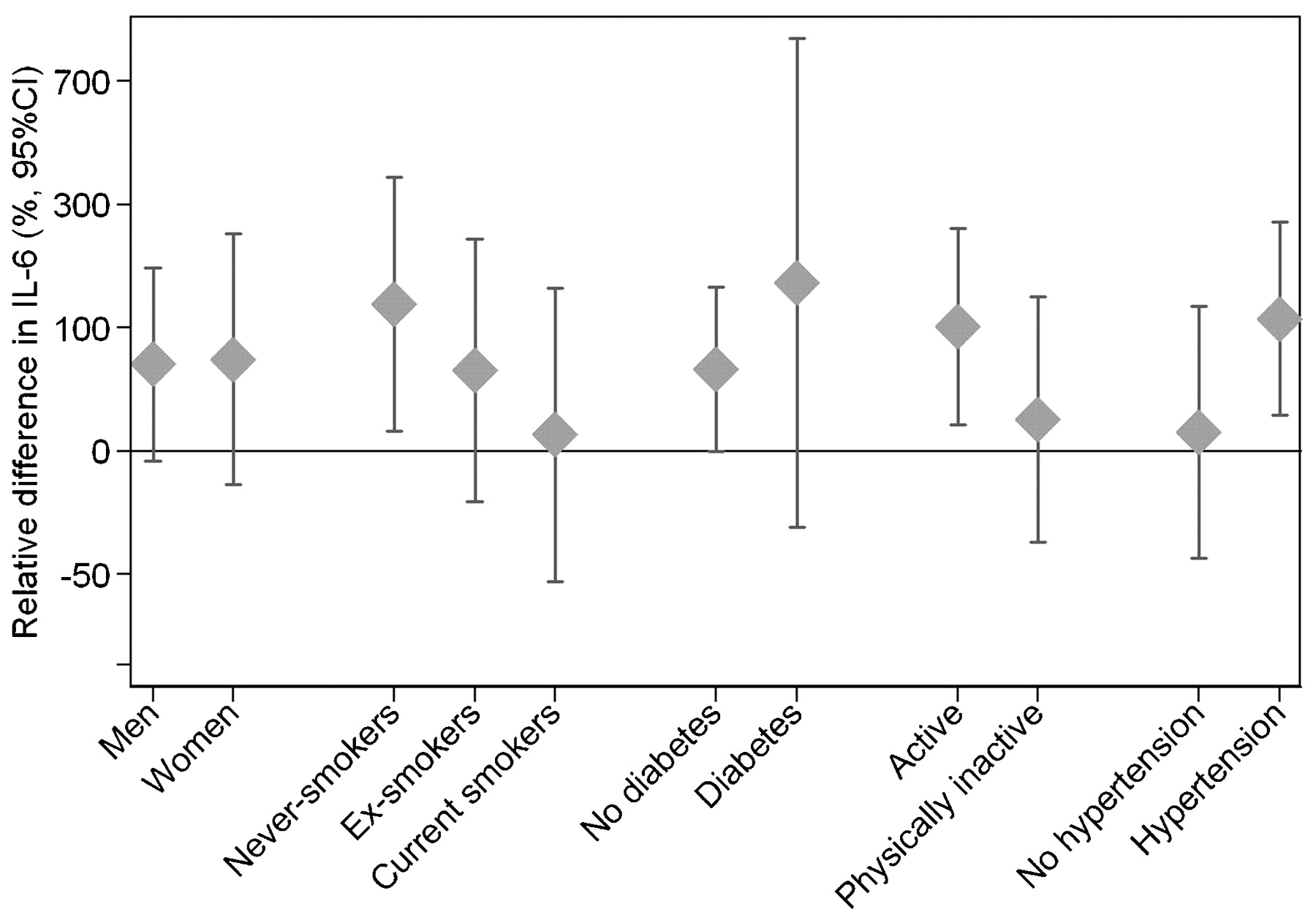{kind=link}
Effect modification of long-term air pollution effects of 30 years exposure to traffic-NO2 on relative IL-6 levels, according to difference in exposure corresponding to the 5th to 95th percentile of individual residential outdoor pollutant levels among subjects in Stockholm (bar indicates 95% confidence interval). Adjusted for age, sex, physical inactivity and the level of HDL cholesterol.
Long-term air pollution effects on blood markers, relative difference (%) in blood marker level according to difference in exposure among subjects in Stockholm
Analysis of the effects of short-term exposure to elevated urban background levels of ambient air pollution on blood markers suggested a possible non-significant trend of increased levels of the inflammatory markers IL-6 and TNF-α related to exposure to NO2 and PM10 0–12 h and 12–24 h before, and CRP levels related to exposure 2 and 5 days before, blood sampling (table 4). For fibrinogen levels, a significant positive association was seen for short-term exposure to O3 during the preceding 2 and 5 days, while a negative association was seen for SO2 exposure. Crude and adjusted effect estimates are presented in online supplementary table S4. A sensitivity analysis adjusting for season indicated very similar effect estimates in relation to NO2 and PM10, while the associations between fibrinogen and O3 and SO2, respectively, were attenuated.
Short-term air pollution effects on blood markers, relative difference (%) in blood marker level according to difference in exposure among subjects in Stockholm*
The analysis restricted to the hospital catchment areas closest to air pollution monitors confirmed the main results for short-term exposure, showing little evidence of improved exposure estimation for this subset of the data.
Discussion
Our findings suggest that air pollution can affect the levels of certain inflammatory markers after both long- and short-term exposures to air pollution. Long-term exposure to source-specific air pollutants in outdoor residential air (over 1, 5 and 30 years) demonstrated a consistent association with increased IL-6 and CRP levels for both traffic-NO2 and heating-SO2. In addition, levels of the inflammatory markers tended to be elevated after short-term exposures to the ambient air pollutants NO2 and PM10. Short-term exposures also seemed to affect the levels of fibrinogen: exposure to O3 demonstrated positive association and SO2 negative association.
Previous epidemiological and experimental studies have presented contradictory results for the effects of short-term exposures to air pollution on levels of various blood markers. Experimental exposure of volunteers to PM resulted in increased fibrinogen levels but showed no association with levels of IL-6, TNF-α, CRP or PAI-1.19 20 In a study exposing men with prior myocardial infarction to diluted diesel exhaust, no changes were detected in the levels of CRP and PAI-1.21 Epidemiological studies have reported inconsistent effects of short-term exposure to ambient air pollutants on the levels of fibrinogen, either with a negative association (in response to PM10 or to welding fume),22 23 a positive association (in response to PM10, NO2, CO or respiratory dust in construction works)18 24 25 26 or no association (in response to PM2.5, PM10, NO2, CO, SO2, O3 or carbon black).24 25 27 28 29 Short-term exposure to particulate matter has been associated with an increase in IL-6 levels18 26 or shown no relationship.22 Similarly, some epidemiological studies of short-term particulate matter exposure reported positive association with CRP levels22 29 or no association.18 28 Based on previous evidence and our results, it is possible that a response to short-term exposure may exist but with considerable variation, resulting in difficulties to consistently detect an effect, whereas continuous exposure may lead to sustained increased levels of inflammatory markers and induction of chronic low-grade inflammation.
The long-term air pollution exposure variables used in our study were designed as indicators for different pollution mixes (traffic-related exhaust or heating-related pollutants). Therefore, it is difficult to disentangle the particular inflammation-inducing agent, given that both gaseous and particulate compounds of air pollution may be capable of inducing an inflammatory response in the lungs.
Our observations of apparent stronger effects of long-term exposure to traffic-NO2 in non-smokers might be explained by the fact that they had no competing influences from tobacco smoke induced low-grade inflammatory responses.30 Thus, the pulmonary inflammatory reactivity to an additional air pollution load may be more difficult to capture in smokers as compared to non-smokers. A higher effect of long-term air pollution exposure on physically active people could be linked to higher pulmonary ventilation and more outdoor exposure, with a consequent higher exposure load of polluted urban air. Other studies have reported exacerbation of asthma symptoms and decrease in lung function in athletes and hikers in response to elevated levels of air pollutants.31 32
In vitro studies have reported increased production of the proinflammatory cytokines TNF-α and IL-6 by alveolar macrophages in response to PM,33 34 35 and in cultured nasal mucosa by exposure to O3 and NO2.36 By inducing the inflammatory cascade, TNF-α stimulates production of other inflammatory molecules, including IL-6.37 In turn, IL-6 induces the production of acute phase proteins, including CRP and fibrinogen.38 Our findings are generally consistent with these mechanisms. With elevated levels of short-term exposure to ambient NO2 and PM10, a tendency for increased production of proinflammatory cytokines and CRP was suggested, implying an immediate response to oxidative damage, albeit of limited magnitude and possibly masked by high variability. The short-term estimates of NO2 are associated with local combustion, which mainly implies traffic-related air pollution in the study area. Short-term variation in PM10 levels is associated with more long-range transboundary transport of air pollutants. The detected positive association of fibrinogen levels with short-term O3 exposure and negative association with short-term SO2 exposure might imply different mechanisms of response to these pollutants. High SO2 levels are associated with the heating season, and O3 is elevated during hot weather periods (the pollutants are negatively correlated; see online supplementary table S2). Also, people in general stay indoors more during particularly cold periods. Consequently, these two results may reflect the same effect of increased fibrinogen with summer-type pollutant mix versus winter-time, although our dataset does not allow a detailed disentanglement of this question. Long-term effects of air pollution exposure, both source-specific traffic-NO2 and heating-SO2, were associated with a significant increase in IL-6 and suggested increase of CRP. This suggests a chronic systemic low-grade inflammation in continuously exposed individuals, with an altered cytokine milieu that could contribute to the development of atherosclerosis,3 ultimately leading to myocardial infarction and other cardiovascular disease manifestations.
Our results were obtained in a large population-based sample with relatively high participation rates. The study subjects were matched to myocardial infarction cases by age, gender and hospital catchment area. This means that the study group is not an entirely representative random sample of the Stockholm population. On the other hand, this selection implies that a higher proportion are elderly and come from more central areas of Stockholm county, where air pollution concentrations are higher and our exposure assessment methodology can be expected to perform better. This should help to improve the power of the study to detect any real effects. Furthermore, we reviewed a large number of factors, including occupational exposures, socio-economic status, dietary patterns and other factors for potential confounding. On the other hand, the assessment of blood marker levels was based only on one reading per subject, and will be affected by measurement error and intra-individual variation related to circadian rhythms, inflammatory conditions, stress levels, etc. In future research, minimising such effects could be achieved by increasing the study population, as well as by performing multiple sampling in the same individual accompanied by information on any relevant characteristics affecting short-term variation.
To our knowledge, this is the first study addressing in the same population the effects on blood markers of both long- and short-term exposure to air pollution. Long-term exposures were assessed using spatial modelling of emissions at each subject’s residential addresses up to 30 years retrospectively, taking changes of residence into account. This method relies on retrospective emission estimates over a long period of time, which can contribute to exposure measurement error. Nevertheless, such misclassification is likely to be non-differential, resulting in attenuation on average of estimated effects. Information on exposure at work, transport and leisure activities was not included in the air pollution exposure measures, but some of the most relevant exposures, such as occupational exposure to diesel and motor exhausts, were assessed separately for their potential confounding effect. No strong confounding was identified by these exposures. Short-term exposure to air pollution was calculated as arithmetic means of hourly inner-city ambient rooftop measurements. This is a crude estimate, because one might expect the pollutant levels to vary over the relatively large geographical study area and, for example, be lower in suburban areas. However, this exposure assessment methodology is commonly used for studies of short-term variation in air pollution levels and its usefulness relies on the assumption that although absolute levels may vary across the study area, the central measurements reflect well the variation in exposure levels in different locations. This argument is stronger for a time-series design, however, and more misclassification of exposure might be expected in a cross-sectional study such as this, but such misclassification of exposure would likely be independent of the measured biomarkers, again resulting in attenuation on average of estimated effects. For a better estimation of short-term air pollution exposure, a more targeted sampling of subjects focusing on the vicinity of monitoring stations as well as modelling of levels more distant from the stations could be the way forward.
Acknowledgments
We thank all SHEEP program participants and the research team, as well as Gunnel Gråbergs and Tomas Lind for their generous help with computer programming.
REFERENCES
Supplementary materials
Web only appendix 66;11:747
Files in this Data Supplement:
Footnotes
Additional tables are published online only at http://oem.bmj.com/content/vol66/issue11
Funding The research was supported by the Swedish Medical Research Council (09533), the Swedish Lung and Heart Foundation, and King Gustaf the V and Queen Victoria’s Foundation.
Competing interests FN is employed by AstraZeneca and AstraZeneca also supports his academic part-time adjunct position as Lecturer in Molecular Epidemiology at Karolinska Institutet. FN also holds some AstraZeneca shares. MR is currently employed by GlaxoSmithKline Biologicals.
Ethics approval The study was approved by the Ethics Committee at Karolinska Institutet.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.