Article Text

Abstract
Objectives To describe clinical characteristics and preoperative management of a national cohort of infants with Hirschsprung's disease (HD).
Design Population-based cohort study of all live-born infants with HD born in the UK and Ireland from October 2010 to September 2012.
Setting All 28 paediatric surgical centres in the UK and Ireland.
Participants 305 infants presenting before 6 months of age with histologically proven HD.
Main outcome measures Incidence, clinical characteristics including gestational age, birth weight, gender, associated anomalies; age and clinical features at presentation; and use of rectal washouts or stoma.
Results The incidence of HD in the UK and Ireland was 1.8 per 10 000 live births (95% CI 1.5 to 1.9). Male to female ratio was 3.3:1. An associated anomaly was identified in 23% (69), with 15% (47) having a recognisable syndrome. The proportion of infants who presented and were diagnosed in the neonatal period was 91.5% (279) and 83.9% (256), respectively. 23.9% (73) and 44.2% (135) passed meconium within 24 and 48 hours of birth. 81% (246) first presented to a hospital without tertiary paediatric surgical services, necessitating interhospital transfer. Initial colonic decompression was by rectal washouts in 86.2% (263) and by defunctioning stoma in 12.8% (39). Subsequently, 27.4% (72) of infants failed management with rectal washouts and required a delayed stoma, resulting in 36.4% (111) of infants having a stoma.
Conclusions In this population-based cohort, presentation outside the neonatal period was rare. Nearly half of the infants with HD passed meconium within 48 hours of birth and over one third were managed with a stoma.
- Hirschsprung's disease
- Incidence
- Associated anomalies
- Management
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
What is already known on this topic?
The estimated incidence of Hirschsprung's disease (HD) is approximately 1 in 5000 live births and more than 90% of cases present in the neonatal period.
Most information on the epidemiology and early management of HD comes from retrospective case series, voluntary reporting surveys and surveys of intended practice.
There is little published information on interactions between infants with HD and healthcare services prior to a diagnosis of HD being established.
What this study adds?
A quarter of infants with Hirschsprung's disease (HD) pass meconium within 24 hours of birth and nearly half within 48 hours of birth, limiting the validity of ‘timing of first meconium’ as a screening question for HD.
One in three infants with HD is discharged home after birth, prior to diagnosis, placing a heavy burden on primary care to ensure timely referral.
More than a third of infants with HD receive a stoma prior to definitive surgery—a higher rate than reported internationally and in contrast to surgeons reported intent.
Introduction
Hirschsprung's disease (HD) is characterised by the absence of intrinsic parasympathetic ganglia (aganglionosis) in the distal bowel, resulting in functional intestinal obstruction. Patients classically present during the neonatal period or early infancy.1 Initial supportive management is followed by definitive surgery, involving resection of the aganglionic colon below the histological ‘transition zone’ (TZ) and anastomosis of ganglionic bowel to the anorectum.2–5
Worldwide, the estimated incidence of HD is approximately 1 in 5000 live births,6 ,7 but there are no nationally representative data to provide an accurate picture of the incidence, demographics and mode of presentation of HD in the UK and Ireland. Available regional data regarding incidence and associated anomalies are limited by a restricted study population, data collection over prolonged periods of time8 or include cases identified more than 50 years ago.9 ,10
Following confirmation of HD, initial management aims to maintain colonic decompression. Most paediatric surgeons advocate rectal washouts to achieve this, aiming to perform a primary pull-through,11 removing the aganglionic bowel without a preceding stoma.12 Some infants fail to decompress adequately with rectal washouts and require a stoma to achieve satisfactory colonic decompression, and some are deemed unsuitable from the outset. The majority of previous studies examining the early management of HD comprise retrospective case series,13 ,14 voluntary reporting surveys with variable methods of case ascertainment,1 ,7 ,15 surveys of intended practice11 ,16 ,17 or meta-analyses and systematic reviews of retrospective case series.18–20 To date, there are very few prospective, population-based observational studies of HD anywhere in the world1 ,7 and none that provide representative data in a cohort of children born during a short time period.
The aims of this study were to describe the incidence, clinical characteristics and management prior to definitive surgery in a national cohort of infants with HD in the UK and Ireland.
Methods
All live-born infants, up to 6 months of age, diagnosed with HD (defined as an absence of ganglia in the enteric nervous system of the distal bowel), between the 1 October 2010 and 30 September 2012 were eligible for inclusion in the study. Cases were identified using the British Association of Paediatric Surgeons Congenital Anomalies Surveillance System.21 Each month, we sent a reporting card to nominated reporting clinicians in all 28 paediatric surgical units in the UK and Ireland, requesting the number of infants diagnosed with HD in their unit that month. In response to a report indicating a new case of HD, we sent a data collection form requesting further details including basic demographic data, age and clinical features at presentation, associated anomalies, early management prior to definitive surgery and site of histopathological TZ. Up to five reminders were sent if the data collection form was not returned.
Duplicate reports were eliminated by comparing hospital of birth, gestation at birth and date of notification and follow-up with the reporting clinicians.
Statistical analyses
Descriptive statistics were used to describe the basic demographics, associated anomalies and mode of presentation. We calculated the rate of HD with 95% CIs among live-born infants by using the denominator of total reported live births in England and Wales,22–24 Scotland,25 Northern Ireland26 and the Ireland27 during the study period, 1 October 2010 to 30 September 2012. All statistical analyses were performed using STATA V.14.
Results
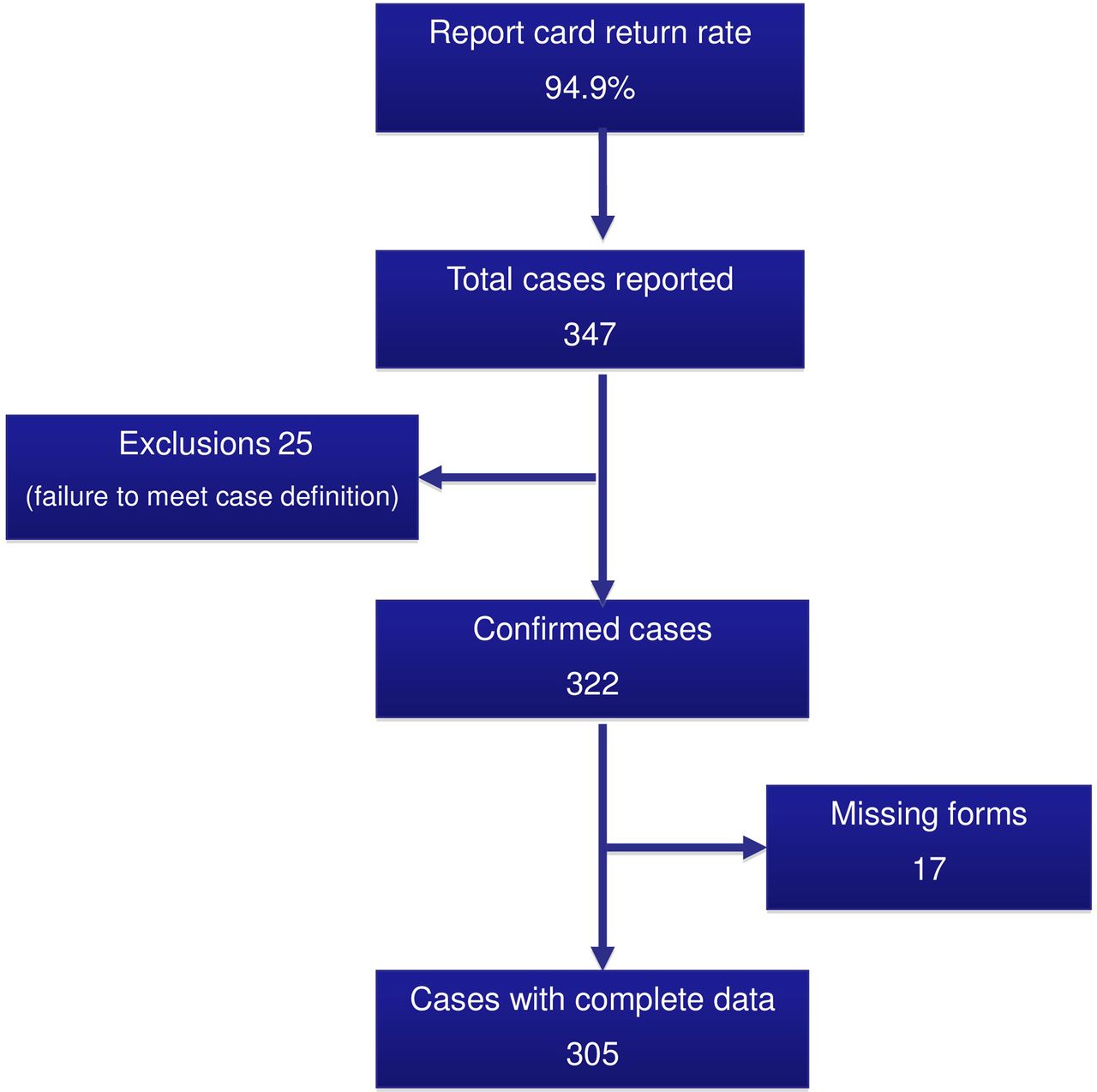
Between 1 October 2010 and 30 September 2012, 305 infants with HD were identified in the UK and Ireland. Figure 1 summarises case ascertainment, exclusions and data collection for the study.

Case ascertainment and data collection.
Incidence
Over the same period, there were 1 729 854 live births in the UK and Ireland.22–27 The incidence of HD in the UK and Ireland was thus estimated as 1.8 per 10 000 live births (95% CI 1.5 to 1.9).
Basic demographics, associated anomalies and mode of presentation
The basic demographics, associated anomalies and presenting features for the cohort are summarised in table 1. Median gestational age was 39 weeks (range 28–42). Thirty-eight infants (12%) were born prematurely (defined as before 37 weeks completed gestation) and 19 infants (6.2%) were born at less than 35 weeks gestation. Median birth weight was 3400 g (range 1000–4900 g). Median age at presentation and diagnosis were 2 (range 1–159) and 9 days (range 1–177), respectively. Median time from presentation to diagnosis was 5 days (range 0–176), and 256 infants (84%) were diagnosed during the neonatal period.
Demographics, associated anomalies and presenting features in 305 infants with Hirschsprung's disease
Overall, the HD cohort included 3.3 times more male than female infants (male 234 vs female 70) and 26 (9%) infants had a positive family history. An associated anomaly was identified in 69 infants (23%), with 47 (15%) of these having a recognisable syndromic association, including Down syndrome (27, 9%), Mowat-Wilson syndrome (5, 2%), congenital central hypoventilation syndrome (3, 1%) or Bardet-Biedl syndrome (2, 1%). An associated cardiac anomaly was identified in 29 infants (10%), which occurred in the context of an underlying predisposition syndrome in 22 out of 47 infants (47%) and in 7 out of 258 non-syndromic infants (3%). An associated urological anomaly was identified in 11 infants (4%), which occurred in the context of an underlying predisposition syndrome in 4 out of 47 infants (9%) and in 7 out of 258 non-syndromic infants (3%).
Presentation
Abdominal distension and bilious vomiting were the most common clinical features at presentation (table 1). In combination, these two features were present in 188 infants (61.6%). Meconium was passed spontaneously within 24 hours of birth in 73 infants (24%) and within 48 hours in 135 infants (44%). The ‘classic triad’ of bilious vomiting, abdominal distension and delayed passage of meconium was evident in only 80 infants (26.2%). The first presentation with symptoms and signs associated with HD occurred in a hospital without tertiary paediatric surgical services in 246 (81%) infants, necessitating transfer to a paediatric surgical centre. One hundred and three (34%) infants presented from home.
Maintenance of colonic decompression
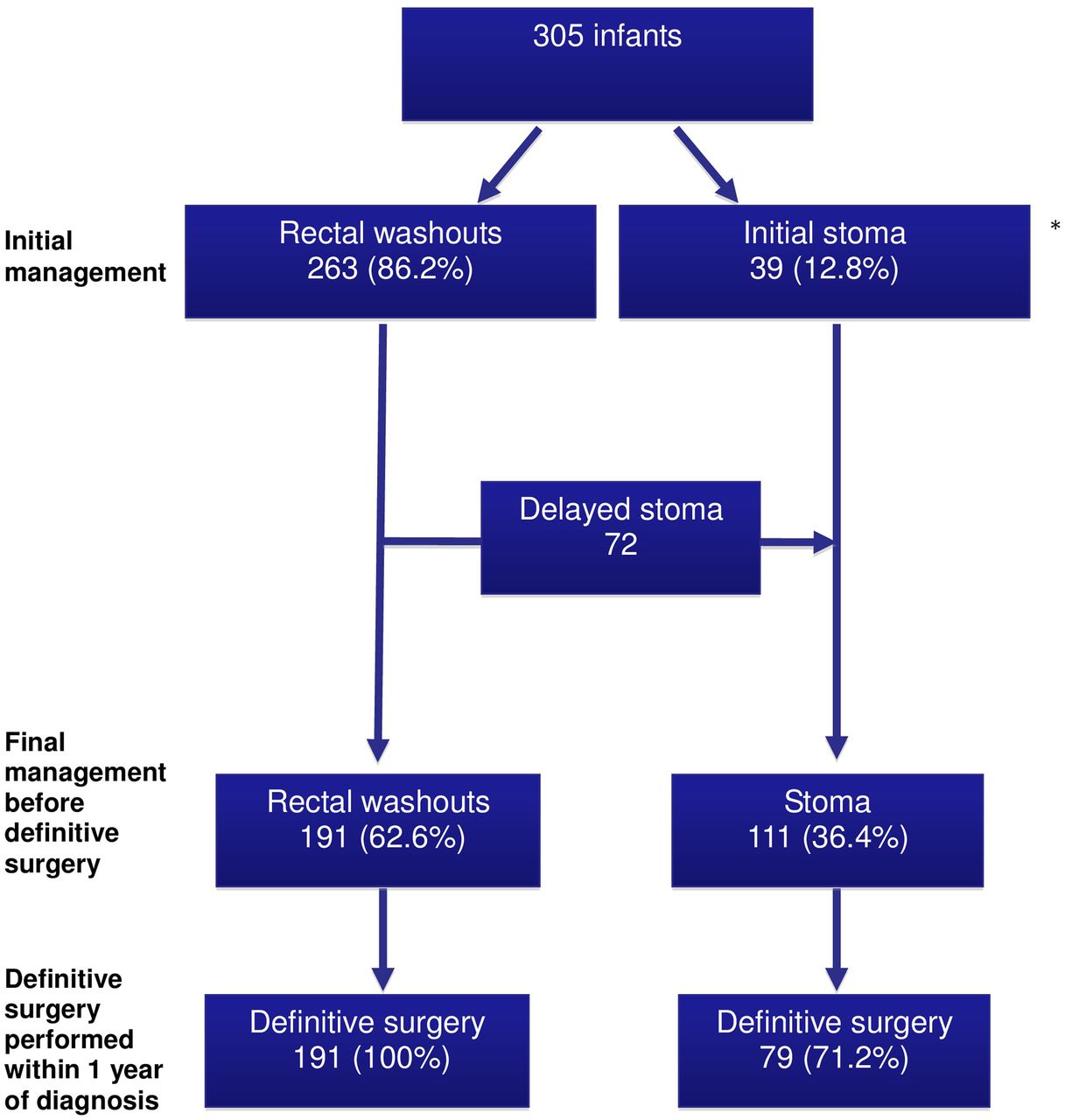
The initial method for colonic decompression (rectal washout vs stoma), the final management prior to definitive surgery and the proportion of infants in each group undergoing definitive surgery within 1 year of diagnosis are summarised in figure 2. Rectal washouts were used in 263 (86%) infants, and 179 (68%) infants had this treatment at home. A stoma was performed in 39 (13%) infants without an initial trial of rectal washouts. One infant died and two underwent a primary pull-through without preceding stoma or rectal washout. A further 72 infants subsequently underwent stoma formation, having initially been managed with rectal washouts. In total, 111 (36%) infants received a stoma prior to definitive surgery, at a median age of 13 days (range 1–367). Indications for stoma formation included failure to decompress with rectal washouts (42), emergency laparotomy (25), suspected long-segment disease (16), enterocolitis (10), consultant preference for a staged approach in all cases (7), comorbidity (7), failure to manage rectal washouts (6) and delayed presentation (1). Following confirmed histological diagnosis, 243 (80%) infants were allowed home before definitive surgery.

{kind=link}
{kind=link}
Maintenance of colonic decompression and the proportion of infants undergoing definitive surgery at 1 year after diagnosis. *Three infants not included—one died and two had a primary pull-through without preceding stoma or rectal washouts.
Length of aganglionosis
Definitive surgery was carried out in 270 (89%) infants within 1 year of diagnosis, establishing the length of aganglionosis. The pathological TZ was rectosigmoid in 198 (73.3%), long segment (proximal to the sigmoid colon) in 60 (22.2%), total colonic in 8 (3.0%) and unknown in 4 (1.5%).
Mortality
Nine infants (3%) died without undergoing definitive surgery. Seven of these infants had a stoma in situ at the time of death and seven had a syndromic association. The median age at death was 85 days (range 11–381). The cause of death was attributed to an underlying cardiac anomaly in five infants, sepsis in two infants, multiple comorbidities prompting withdrawal of treatment in one infant and was unclear in one infant.
Discussion
This study provides robust, population-level data for the UK and Ireland, with data collected directly from local surgeons, rather than ‘second hand’ from administrative databases. The population-based nature, together with high rates of case ascertainment and data accrual achieved, allow an accurate estimate of the incidence, clinical characteristics and current preoperative management of infants with HD in the UK and Ireland.
To enable future comparative, age-matched analysis of long-term functional outcome, we limited our cohort to infants diagnosed before 6 months of age. Contemporary population-based studies confirm that around 90% of infants with HD are diagnosed within the neonatal period1 and late-presenting cases are rare.28 While it is likely that a small number of cases presenting after 6 months of age will not have been captured, we feel that this will have little impact on incidence calculations and that our cohort is representative of the vast majority of children with HD in the UK and Ireland.
For a condition with a complex pattern of polygenic inheritance, characterised by variable sex-dependent penetrance of the most common known genetic mutations,29 the incidence of HD worldwide appears consistent with the caveat that most studies originate from the Caucasian diaspora.1 ,6 ,7 In the UK and Ireland, two small, non-population-based studies from the 1980s and 1960s, respectively, estimated the incidence of HD to be 1 in 4500 live births9 and 1 in 2000–10 000 live births,10 but both studies were limited, either by a protracted study period9 or through collection of cases from a wide geographical area, with a poorly defined study population.10 The incidence of 1.8 per 10 000 live births calculated from our study is comparable to the incidence of 1.63 per 10 000 live births (95% CI 1.33 to 1.98) identified in the North of England using the Northern Congenital Abnormality Survey between 1990 and 2008.8 A recent European Surveillance of Congenital Anomalies study covering 31% of the European birth population between 1980 and 200930 found a total prevalence of 1.09 per 10 000 live births (95% CI 1.03 to 1.15) with marked regional variation in prevalence. The authors concede that this may result from differences in case definition and heterogeneity in regional reporting rates, which may also account for the lower than expected incidence compared with the majority of published series.1 ,6 ,7
In our cohort, 12% of infants with HD were born prematurely; nearly twice the overall rate of preterm birth for England and Wales (7.3%)31 and Scotland (5.9%)32 during the study period. A recent systematic review19 reported a 6% rate of preterm birth in HD and suggested a rising prevalence. Baxter and Bhatia33 suggest that the observed rise in prevalence of prematurity in HD may be attributable to a rising incidence of prematurity overall, but since the population incidence of prematurity has remained at 7.3% for England and Wales between 2009 and 2012,31 and has fallen from 6.7% in 2003/2004 to 5.9% in 2011/2012 in Scotland,32 other factors should be considered. Population-based birth defect surveillance systems have previously identified an association between other major birth defects and preterm birth.34 Far from being rare in infants with HD, there is growing evidence of an association between HD and preterm birth, and future work should seek to clarify this potential relationship.
In our cohort, more than one in five infants had an associated anomaly, with a syndromic association in 15% and an isolated additional anomaly in 7%. Overall, 1 in 11 infants had Down syndrome and this group accounted for 57.4% of infants with a predisposing syndrome. Infants with Down syndrome are estimated to have 40 times increased risk of HD.35 This was confirmed by a recent meta-analysis of more than 16 000 infants with HD, which found a 7.3% rate of Down syndrome, compared with an expected rate of 0.15%–0.17% in the general population.20 The finding that HD occurred as an isolated trait in 77.4% of infants in this series is in agreement with previous studies.29
Between 94%36 and 98.5%37 of normal-term infants pass meconium within 24 hours of delivery and the remainder by 48 hours.37 Previously, it has been suggested that less than 10% of infants with HD pass meconium within 24 hours of birth.38 In this study, a quarter of infants with HD passed meconium within 24 hours of birth and nearly half of infants within 48 hours of birth. Two recent studies1 ,13 also reported that around 40% of infants with HD, including preterm infants,13 passed meconium within 48 hours of birth. Current National Institute for Health and Care Excellence (NICE) guidance on ‘Constipation in Children and Young People’ incorporates ‘delayed passage of meconium beyond 48 hours in term infants’ as a red flag for urgent specialist referral to exclude HD.39 Our findings suggest that the apparently timely passage of meconium should not lead the clinician to refute a diagnosis of HD, particularly in the presence of other ‘red flag’ symptoms such as abdominal distension.
Although paediatric surgeons are familiar with the management of HD, our findings indicate that the majority of these infants initially present to other primary or secondary healthcare services in the UK and Ireland. One third of infants in this cohort were discharged home after birth, prior to HD being suspected or diagnosed and over 80% first presented to a hospital without tertiary paediatric surgical services and required transfer to a paediatric surgical centre. As the majority of these infants presented initially to non-surgical specialities, these data highlight the importance of regional networking, including robust referral pathways to tertiary care services.
Surveys of practice have demonstrated clear changes in surgeons' preference for the initial colonic decompression of infants with HD, with a move away from a staged approach and increased popularity of the primary pull-through.11 ,15 ,16 In a recent survey of intended practice of UK paediatric surgeons, only 15% would aim to perform a stoma prior to definitive surgery.11 In the presented cohort, a defunctioning stoma was actually performed in more than one third of infants, with 13% receiving a stoma without any attempts at rectal washouts, and a further 27% of infants initially managed with rectal washouts receiving a stoma, after the washouts failed to achieve adequate decompression. These findings provide robust information that can be used to counsel parents of infants with HD. Limited data exist to explain the higher than expected stoma rates observed in clinical practice compared with surveys of intended practice. In this cohort, the indications for stoma formation were mostly due to clinical concerns, rather than a consultant preference for this approach. Future work will be directed at identifying factors that increase the likelihood of stoma formation.
Conclusions
This study identified a national cohort of infants with HD in the UK and Ireland. The data provide a robust estimate of the incidence, clinical characteristics and associated anomalies of HD in the UK and Ireland. We report national outcome data for clinical markers such as time from presentation to diagnosis and stoma utilisation rates that can be used as a benchmark against which practice and outcomes in single centres can be compared and future changes in service provision measured. The data provide further evidence to challenge some of the previously held dogmas in HD, including the prevalence of preterm birth and the validity of timing of first meconium as a screening question for HD. There appears to be an increased use of home rectal irrigations and high stoma rates, both of which rely on adequate support services in the community. Furthermore, one in three infants with HD is discharged home after birth, prior to a diagnosis of HD, placing a heavy burden of responsibility on health visitors and general practitioners to ensure timely referral of these infants.
Acknowledgments
All individuals who meet the ICMJE criteria have been included in the list of authors. This study, however, would not have been possible without the collaboration of Elizabeth Draper, Paul Johnson and Jennifer J Kurinczuk who assisted with development of the funding application, and the BAPS-CASS reporting clinicians throughout the UK and Ireland. We also thank Patsy Spark, Melanie O'Connor and Anne Smith from the National Perinatal Epidemiology Unit who helped with generating the data collection forms, programming and data entry.
References
Footnotes
Collaborators Elizabeth Draper, Adil Aslam, Marcin Kazmierski, Wajid Jawaid, Ingo Jester, Anindya Niyogi, Eleri Cusick, Janet McNally, Simon Clarke, Julie Galea, Shabnam Parker, Emily Broadis, Ewan Brownlee, Charles Keys, Dorothy Kufeji, Nicholas Alexander, Jo Curry, Samir Gupta, Saravanakumar Paramalingam, Sanja Besarovic, Paul Johnson, Ceri Jones, Bhanu Lakshminarayanan, Helen Carnaghan, Shailesh Patel, Ian Sugarman, Shawqui Nour, Thomas Tsang, Jenny Kurinczuk, Prem Puri, Suzanne McMahon, Sandeep Motiwale, Shailinder Singh, Richard Thompson, Costa Healy, Anies Mahomed, David Marshall, Evelyn Ervine, Lara Shipley, Augusto Zani, Paul Charlesworth, Ashwini Joshi, Simone Ragazzi, Ross Craigie, Sumita Chhabra, Bruce Jaffray, Anne Lawson, Sean Marven, Karen Lloyd, Govind Murthi, Michael Stanton, Simon Blackburn, Stefano Giuliani, Eric Nicholls, Bruce Okoye, Dermot McDowell, Alan Mortell, Suzanne McMahon, Simon Huddart, Ram Shrestha, Julianne Rowsell on behalf of the British Association of Paediatric Surgeons Congenital Anomalies Surveillance System.
Contributors TJB designed the study protocol and data collection forms, monitored data collection for the whole study, wrote the statistical analysis plan, cleaned and analysed the data and drafted and revised the paper. MK designed the study protocol and data collection forms, monitored data collection for the whole study, contributed to the statistical analysis plan, helped analyse the data and revised the paper. SK helped design the study protocol and data collection forms; contributed to the statistical analysis plan and revised the paper. MN wrote the statistical analysis plan, cleaned and analysed the data and revised the paper. GMW designed the study protocol and data collection forms, monitored data collection for the whole study, wrote the statistical analysis plan and drafted and revised the paper. All authors had full access to all of the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. GMW is guarantor. GMW affirms that the manuscript is an honest, accurate and transparent account of the study being reported, that no important aspects of the study have been omitted and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
Funding This study was funded by the charity BDF Newlife and the British Association of Paediatric Surgeons. MK is funded by an NIHR Research Professorship (NIHR-RP-011-032). The views expressed in this publication are those of the author(s), and not necessarily those of the NHS, the NIHR, or the Department of Health. All authors are independent of the funders.
Competing interests None declared.
Ethics approval This study was approved by the West of Scotland REC 4 (Ref: 10/S0704/47).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data sharing is governed by the National Perinatal Epidemiology Unit Data Sharing Policy, which can be obtained from the corresponding author.