Article Text

Abstract
Objectives Interleukin 33 (IL-33) is a new member of the IL-1 family of cytokines which signals via its receptor, ST2 (IL-33R), and has an important role in Th2 and mast cell responses. This study shows that IL-33 orchestrates neutrophil migration in arthritis.
Methods and results Methylated bovine serum albumin (mBSA) challenge in the knee joint of mBSA-immunised mice induced local neutrophil migration accompanied by increased IL-33R and IL-33 mRNA expression. Cell migration was inhibited by systemic and local treatments with soluble (s)IL-33R, an IL-33 decoy receptor, and was not evident in IL-33R-deficient mice. IL-33 injection also induced IL-33R-dependent neutrophil migration. Antigen- and IL-33-induced neutrophil migration in the joint was dependent on CXCL1, CCL3, tumour necrosis factor α (TNFα) and IL-1β synthesis. Synovial tissue, macrophages and activated neutrophils expressed IL-33R. IL-33 induces neutrophil migration by activating macrophages to produce chemokines and cytokines and by directly acting on neutrophils. Importantly, neutrophils from patients with rheumatoid arthritis successfully treated with anti-TNFα antibody (infliximab) expressed significantly lower levels of IL-33R than patients treated with methotrexate alone. Only neutrophils from patients treated with methotrexate alone or from normal donors stimulated with TNFα responded to IL-33 in chemotaxis.
Conclusions These results suggest that suppression of IL-33R expression in neutrophils, preventing IL-33-induced neutrophil migration, may be an important mechanism of anti-TNFα therapy of inflammation.
Statistics from Altmetric.com
Introduction
The interleukin 1 (IL-1) family of cytokines includes 11 members classified systematically as IL-1F1 to IL-1F11.1 IL-33 is the latest member of the family and is also known as IL-1F11.1 2 IL-33 is structurally related to IL-1β and IL-18 and was found to have strong biological relevance in immune and inflammatory reactions.2 However, unlike IL-1β and IL-18, which mainly promote Th1-/Th17-associated responses, IL-33 enhanced IL-5 and IL-13 production by polarised Th2.2 3 In addition, administration of IL-33 into naïve mice provoked innate type II cytokine and IgE production.2 IL-33 signals via a heterodimeric receptor—the α-chain IL-33R2 and the β-chain IL-1RAcP4 5—and triggers the activation of NFκB, MAP kinases p38, ERK1/2, JNK1/2 in mast cells2 and T cells.3
There are two isoforms of the IL-33R protein encoded by the st2 gene and resultant alternative gene splicing: IL-33RL, a transmembrane form, and soluble IL-33R (sIL-33R),6 a secreted form which can serve as a decoy receptor for IL-33.2 7 IL-33RL is preferentially expressed on a subset of Th2 cells but not Th1 cells8 9 and can profoundly modulate innate and adaptive immunity.10 11 IL-33R is also expressed on mast cells, macrophages and fibroblasts.2 Soluble IL-33R is a potent inhibitor of collagen-induced arthritis (CIA) in mice,7 suggesting that IL-33–IL-33R signalling is a key pathway in the context of rheumatoid arthritis (RA). We have also shown that IL-33 contributes to the development of local inflammatory cutaneous and articular mechanical hypernociception (inflammatory pain) in a murine model of RA via interferon γ (IFNγ) production in vivo.12 IL-33 can also induce IFNγ production by Th1 lymphocytes, invariant natural killer-like T (NKT) and natural killer (NK) cells.13 14 As neutrophils contribute importantly to the development of pain,15,–,18 inflammation and tissue destruction in RA,19,–,22 we investigated whether IL-33 has a role in arthritis by enhancing neutrophil migration to inflamed joints.
Materials and methods
Details are given in the online supplement.
Mice
BALB/c, C57BL/6, IL-33R–/–, TNFR1–/–, CCL3–/– and mast cell-deficient (WBB6F1–w/w (v)) mice were bred and maintained in the Faculty of Medicine of Ribeirao Preto (FMRP).
Clinical samples
Peripheral blood samples were collected from eight healthy volunteers, four patients treated with methotrexate (MTX) and nine treated with MTX plus anti-tumour necrosis factor (anti-TNF). Neutrophils were purified and IL-33R expression was determined by quantitative PCR (qPCR) before or after culturing with TNFα. Purified neutrophils were also examined for their chemotaxis to IL-33 in a Boyden chamber.
Immunisation and neutrophil migration
Mice were immunised with methylated bovine serum albumin (mBSA) in complete Freund's adjuvant (CFA). Sham immunised mice received CFA only. Mice were challenged with mBSA or IL-33 on day 21.12 Knee joints cells were harvested, the total number of infiltrating cells was determined in Neubauer chamber and differential counts were performed in slices stained by the Rosenfelt method.23
Macrophages
Thioglycolate-elicited peritoneal macrophages were harvested and then analysed by qPCR or stimulated in vitro with IL-33 followed by cytokine measurement in the supernatant. Some mice were injected intraperitoneally with thioglycolate or saline24 and then injected intraperitoneally 4 days later with IL-33 and neutrophil counts were determined.
Neutrophil isolation and chemotaxis
Neutrophils were isolated by Percoll gradient from femur bone marrow of mice or human peripheral blood.25 The purity of the neutrophils was >98%. qPCR analysis12 or chemotaxis in a Boyden chamber was performed. Some neutrophils were also cultured with TNFα and then assayed for chemotaxis or IL-33R expression by qPCR.
Synovial tissue isolation
Synovial tissue was collected from the knee joints of immunised mice26 and, after culture with IL-33, qPCR analysis or TNFα measurement was performed.
qPCR
Mice β-actin, IL-33 and IL-33R primers were as previously described.12 Human primers are shown in the online supplement.
Proteoglycan quantification assay
Chondroitin sulfate from patella samples was measured using 1,9-dimethyl-methylene blue.27
Statistical analysis
The results are presented as mean±SEM of two independent experiments. p<0.05 was considered statistically significant.
Results
IL-33 mediates mBSA-induced neutrophil migration to the knee joint
Antigen-induced arthritis (AIA) exhibits delayed-type hypersensitivity and the histopathology closely resembles that observed in human RA.28 The intra-articular challenge with mBSA induced dose-dependent and time-dependent neutrophil migration into the knee joint which peaked with 10 µg mBSA/joint and at 24 h after challenge (figure 1A and B). This dose and time were used in all subsequent experiments.

Interleukin 33 (IL-33) mediates methylated bovine serum albumin (mBSA) challenge-induced neutrophil migration to the knee joint of mice. (A) Knee joint cells were harvested 24 h after mBSA challenge (1–10 µg/ joint) or saline injection. (B) Knee joint cells were harvested 6–48 h after mBSA challenge (10 µg/ joint) or saline injection. (C) Mice were treated with soluble (s)IL-33R (100 µg/intraperitoneally/mice/30 min) or Fc control before mBSA (10 µg/joint) challenge. Cells were harvested 24 h later. (D) Mice were treated with intra-articular sIL-33R (3 µg/mice/co-injection with mBSA) or Fc control before mBSA (10 µg/ joint) challenge. Cells were harvested 24 h later. (E) IL-33R+/+ (Balb/c background) and IL-33R–/– (Balb/c background) mice were challenged with mBSA (10 µg/joint) or saline. Cells were harvested 24 h later. (F, G) Knee joint samples were harvested at indicated times and processed for IL-33 (F) or IL-33R (G) mRNA expression determination by quantitative PCR. (H) Knee joint cells were harvested 6 h after IL-33 challenge (30–1000 ng/joint) or saline injection. (I) Knee joint cells were harvested 6–48 h after IL-33 challenge (300 ng/joint) or saline injection. (J) IL-33 (300 ng/joint) was injected into mice pretreated with sIL-33R or Fc control (100 µg/mice/intraperitoneally/30 min). (K) IL-33R+/+ (Balb/c background) and IL-33R–/– (Balb/c background) mice were challenged with saline, mBSA (10 µg/joint) or IL-33 (300 ng/joint) at days 1 and 5. Ten days after the first challenge, knee joint samples were harvested and processed for analysis of proteoglycan content. All mice were immunised against mBSA unless otherwise stated. Data are mean±SEM, n=8 per experiment, representative of two separate experiments. *p<0.05 vs saline control group; †p<0.05 vs 6 h (panel B), Fc control (panel C–E and J), IL-33 dose of 30 ng (panel H), 1 h (panel I) or respective IL-33R+/+ group (panel K). ‡p<0.05 compared to the dose of 1 µg of mBSA (panel A).
We first tested the role of IL-33 in local neutrophil influx in AIA by pretreating the mice with sIL-33R (100 µg/mouse), the decoy receptor of IL-33. Systemic (intraperitoneal) and local (intra-articular, co-injection with mBSA) treatment with sIL-33R significantly inhibited neutrophil migration in AIA (figure 1C and D), suggesting that IL-33 may have an important role in neutrophil activity in this model of inflammatory disease. Confirming this pharmacological approach, IL-33R–/– mice also showed reduced neutrophil migration to the knee joint compared with wild type mice (Balb/c background) (figure 1E). In agreement, mBSA challenge of immunised mice induced an early increase in IL-33 (figure 1F) and IL-33R (figure 1G) mRNA expression.
We next investigated whether IL-33 could mimic mBSA challenge-induced neutrophil migration in vivo. Recombinant IL-33 (30–1000 ng) was injected into the joints of mice and the number of neutrophils in the joints was determined. IL-33 induced a significant local influx of neutrophils in a dose-dependent and time-dependent manner (figure 1H and I). Furthermore, the IL-33-induced neutrophil influx was markedly prevented by the presence of sIL-33R (100 µg/mouse), demonstrating the specificity of IL-33-induced cell migration (figure 1J). Further supporting a role for IL-33 in AIA inflammation and cartilage destruction, the mBSA (10 µg) challenge-induced reduction of proteoglycan content in the joint was absent in IL-33R–/– mice (figure 1K). Similar to mBSA challenge, IL-33 (300 ng) also induced joint proteoglycan loss in an IL-33R-dependent manner (figure 1K).
IL-33-induced neutrophil migration is dependent on CXCL1, CCL3, TNFα and IL-1β
Cytokines may induce neutrophil migration through further production of other cytokines and chemokines.29,–,31 CXCL1, CCL3, TNFα and IL-1β are some of the important chemokines/cytokines for neutrophil recruitment.29,–,31 We therefore investigated the role of these chemokines/cytokines in mediating neutrophil recruitment induced by AIA and IL-33 (see figure S1 in online supplement). Joint tissues from wild type mice immunised and challenged with mBSA (10 µg) contained significant amounts of CXCL1, CCL3, TNFα and IL-1β compared with that of control mice. This increase in cytokine/chemokine production was significantly reduced in IL-33R–/– mice. Conversely, neutrophil influx into the joints in AIA mice challenged with mBSA (10 µg) or in IL-33 (300 ng)-injected mice was markedly reduced when the mice were treated with anti-CXCL1 antibody (700 ng, co-injection) or IL-1ra (30 mg/kg/intravenously/15 min before challenge plus 21 h after challenge for mBSA challenge or one treatment 15 min before challenge for IL-33 stimulus), or in mice deficient in CCL3 or TNFR1. These results therefore show that IL-33-mediated neutrophil migration during specific antigen challenge is dependent on at least the following downstream mediators: TNFα, IL-1, CXCL1 and CCL3.
Association of IL-33 and other mediators in the induction of neutrophil migration
In several models of inflammation29,–,33 neutrophil migration is mediated by a network of different mediators which may act in parallel, in synergy or in sequence. We therefore determined the association of IL-33 with CXCL1, CCL3, TNFα and IL-1β. Corroborating the data in figure S1 in the online supplement, IL-33 injection in vivo induced significant CXCL1, CCL3, TNFα and IL-1β. The IL-33-induced production of these cytokines and chemokines was modulated in IL-33R–/–, CCL3–/–, TNFR1–/– mice or in IL-1ra-treated mice (see figure S2 in online supplement), suggesting that these cytokines and chemokines appear to act in a cascade manner since IL-33-induced TNFα production was not affected by targeting IL-1β, CCL3 or CXCL1 which regulate the production of each other.
IL-33 induces neutrophil migration by activating synoviocytes and macrophages
Mast cells express IL-33R and respond to IL-33.2 4 34 We therefore investigated the potential role of mast cells in IL-33-induced neutrophil migration. Intra-articular injection of IL-33 (300 ng) induced similar levels of neutrophil influx in the joints of wild type and mast cell-deficient mice (figure 2A), which shows that, although IL-33 can activate mast cells,3 4 34 the IL-33-induced neutrophil migration is independent of the mast cells. In contrast, synovial tissue contains significant numbers of fibroblasts and macrophages26 which express IL-33R (figure 2B) and IL-33 (figure 2C). Peritoneal macrophages also express IL-33R (figure 2B) and IL-33 (figure 2C). We therefore investigated the ability of IL-33 to activate synoviocytes and peritoneal macrophages to produce proinflammatory cytokines and chemokines. Synovial tissue activated with IL-33 produced significantly more TNFα than cells stimulated with medium alone (figure 2D). Moreover, macrophages cultured with IL-33 (8–210 ng/ml) produced increased levels of CXCL1, CCL3, TNFα and IL-1β in a concentration-dependent manner (figure 2E–H).
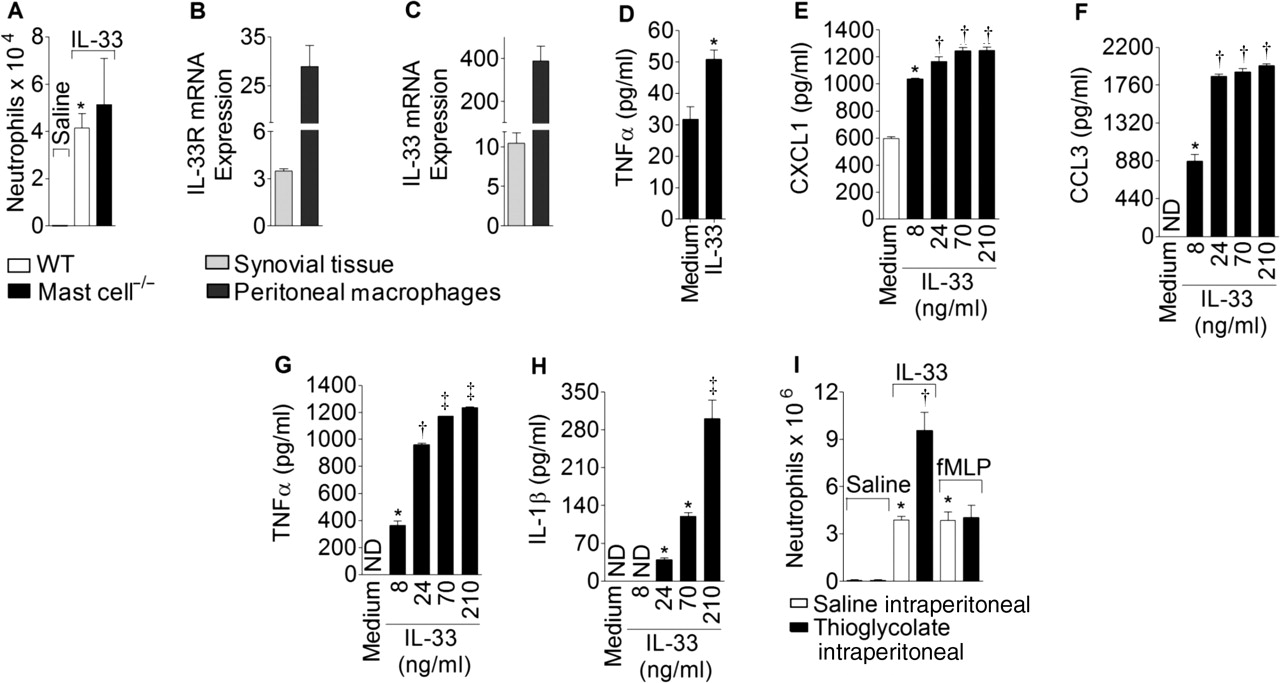
Interleukin 33 (IL-33) induces neutrophil migration by activating synoviocytes and macrophages. (A) Wild type (WT) and mast cell–/– mice received intra-articular injection of IL-33 (300 ng/joint). (B, C) Quantitative PCR analysis for IL-33R (B) or IL-33 (C) mRNA expression by synovial tissue and macrophages. (D) Synovial tissue was stimulated with IL-33 (70 ng/ml/4 h). Supernatant was assayed for tumour necrosis factor α (TNFα) by ELISA. (E–H) Macrophages were stimulated with graded concentrations of IL-33 (8–210 ng/ml/4 h). Supernatant was assayed for CXCL1 (E), CCL3 (F), TNFα (G) and IL-1β (H). (I) Mice were challenged intraperitoneally with IL-33 (30 ng) or N-formyl-methionyl-leucyl-phenylalanine (fMLP) (25 nmol) 4 days after intraperitoneal injection with thioglycollate or saline. Peritoneal cells were harvested for neutrophil counts 6 h after stimulus. Data are mean±SEM, n=5–6 per experiment, representative of two separate experiments. *p<0.05 vs saline or medium control group, †p<0.05 vs saline intraperitoneal injection (open bars, panel I), IL-33, 8 ng/ml (panels D–G); ‡p<0.05 vs IL-33, 24 ng/ml (panel G) and 70 ng/ml (panel H).
To confirm the role of macrophages in mediating the effects of IL-33, experiments were conducted in a classical model of inflammation24 in which thioglycolate was injected into the peritoneal cavity before the injection of the neutrophil-inducing stimuli. Thioglycolate induces a 2–3-fold increase in the number of macrophages in the peritoneal cavity on day 4 after injection.24 IL-33 (300 ng) administration induced a further significant increase in neutrophil influx into the peritoneal cavity (figure 2I). In contrast, the administration of N-formyl-methionyl-leucyl-phenylalanine (fMLP, 25 nmol) did not affect the influx of neutrophils (figure 2I). fMLP is known to induce neutrophil migration directly and independently of macrophage activation.24 These experiments were carried out in the peritoneal cavity instead of the knee joint because of technical difficulties associated with multiple injections into the knee and were used by others for a similar purpose.19 24 Together, these results indicate that macrophages could be a cell type in the synovial tissue responding to IL-33.
IL-33 is also a direct chemoattractant for neutrophils
A series of experiments was then conducted to investigate whether IL-33 could also have a direct effect on neutrophils. Neutrophils were purified from the bone marrow of naïve (naïve neutrophils) or mBSA-immunised mice. The cells were tested for their ability to migrate in response to IL-33 in vitro in a Boyden chamber. Naïve neutrophils did not migrate in response to IL-33 (10–100 ng/ml) but migrated positively to fMLP (10−7 M) and CXCL1 (30 ng/ml) (figure 3A). In contrast, IL-33 induced significant chemotaxis of bone marrow neutrophils from mBSA-immunised mice (figure 3A). Moreover, the chemoattractant effect of IL-33 (100 ng/ml) was observed with neutrophils of immunised IL-33R+/+ mice but not immunised IL-33R–/– mice (figure 3A). Neutrophils from naïve or immunised mice migrated equally well in response to fMLP (10−7 M) or CXCL1 (30 ng/ml) (figure 3A). IL-33 attracted neutrophils only when it was added to the bottom of the chamber but not when added with the neutrophils (top) or to both compartments (figure 3B), indicating that IL-33 induced neutrophil chemotaxis and not chemo- kinesis.35 Consistent with this finding, IL-33R mRNA levels were significantly higher in bone marrow neutrophils from mBSA-immunised mice than from naïve mice (figure 3C).

Chemotactic activity of interleukin 33 (IL-33) on mice neutrophils. (A) Neutrophils from naïve or methylated bovine serum albumin-immunised IL-33R+/+ or IL-33R–/– mice were added in the top compartment and IL-33 (10–100 ng/ml), N-formyl-methionyl-leucyl-phenylalanine (fMLP) (10−7 M) or CXCL1 (30 ng/ml) were added in the bottom compartment of a Boyden chamber. (B) Neutrophils were added in the top compartment and IL-33 (100 ng/ml) was added in the top and/or bottom chamber. (C) Quantitative PCR (qPCR) analysis of IL-33R mRNA expression by neutrophils. (D) Neutrophils from naïve mice were treated in vitro with medium only or tumour necrosis factor α (TNFα) (100 ng/ml) for 2 h, then neutrophils were added in the top compartment and IL-33 (100 ng/ml) in the bottom chamber. (E) TNFα-treated and TNFα-untreated neutrophils were added in the top compartment and fMLP (10−7 M) was added in the bottom chamber. (F) qPCR analysis of IL-33R mRNA expression by neutrophils. (G–I) Neutrophils of naïve mice or immunised TNFR1+/+ and TNFR1–/– (all C57BL/6 background) were added in the top chamber. Medium, IL-33 (100 ng/ml) (G) or fMLP (10−7 M) (H) were added in the bottom chamber. (I) qPCR analysis of IL-33R mRNA expression by neutrophils. All neutrophils were harvested from the bone marrow of mice. Data are mean±SEM, samples were pooled from 12 mice per group, representative of two separate experiments. *p<0.05 vs medium only or naïve neutrophils; †p<0.05 vs lower concentrations of IL-33 (10 and 30 ng/ml) (panel A), IL-33 bottom (panel B), naïve neutrophil+medium pretreatment (panel F) and TNFR1+/+ mice (panels G and I); ‡p<0.05 vs neutrophils of IL-33R+/+ mice (panel A).
We next evaluated whether cytokines that are important in the pathogenesis of RA can influence the activity of the chemotatic effect of IL-33. Neutrophils from naïve mice were incubated with medium alone or with TNFα (100 ng/ml, 2 h). Naïve neutrophils treated with TNFα were responsive to chemoattraction by IL-33 (figure 3D). This effect was selective for IL-33 since the chemoattraction by other direct chemoattractants such as fMLP was similar for neutrophils treated or untreated with TNFα (figure 3E). To further support the notion that TNFα makes neutrophils responsive to IL-33 and that this event occurs during disease, we assessed the chemotatic effect of IL-33 on neutrophils from naïve and immunised TNFR1+/+ and TNFR1–/– mice (C57BL/6 background) (figure 3F, G). Similar to the in vitro effect of TNFα, only neutrophils of immunised TNFR1+/+ mice were chemoattracted by IL-33, but not those of naïve or TNFR1–/– mice (figure 3F). All three groups responded equally to fMLP (figure 3G).
Chemoattraction of neutrophils from patients with RA
The effect of IL-33 on neutrophils of peripheral blood of healthy donors (no clinical sign of disease on anamnesis or clinical investigation) and patients with RA was then investigated. All 13 patients with RA fulfilled the 1987 revised criteria of the American College of Rheumatology for the diagnosis of RA.36 37 Four of the patients were receiving treatment with MTX alone and nine were receiving treatment with MTX plus anti-TNF (infliximab) (see table S1 in online supplement). Neutrophils from the peripheral blood of patients treated with MTX alone migrated in response to IL-33 (100 ng/ml) assessed in the Boyden chamber (figure 4A). In contrast, neutrophils from patients with RA treated with MTX plus anti-TNF or healthy donors did not migrate in response to IL-33 (figure 4A). Consistent with this finding, neutrophils from patients with RA treated with MTX alone expressed higher levels of IL-33R mRNA than neutrophils from healthy donors or from patients with RA treated with MTX plus anti-TNFα (figure 4B). Neutrophils from all three groups of donors migrated similarly to fMLP (10−7 M) (figure 4C). Furthermore, pretreatment of peripheral blood neutrophils from healthy donors with TNFα (100 ng/ml) increased IL-33R mRNA expression (figure 4D) and rendered these cells responsive to chemoattraction by IL-33 (figure 4E). TNFα-treated and TNFα-untreated neutrophils from healthy donors migrated equally to fMLP (figure 4F). These data therefore show that TNFα can contribute to local inflammation by inducing IL-33R expression on neutrophils which then migrate to the site of inflammation in response to IL-33 produced by a range of cell types within the synovium. Furthermore, these results also indicate that a reduction of IL-33R expression and hence in neutrophil migration to the joints may be an important mechanism for the effective therapy of TNF targeting in RA.

Chemotactic activity of interleukin 33 (IL-33) on human neutrophils. (A) Human neutrophils were added in the top compartment and IL-33 (100 ng/ml) was added in the bottom and/or top compartment of the Boyden chamber. (B) Quantitative PCR (qPCR) analysis of IL-33R mRNA expression by neutrophils from human peripheral blood. (C) Human neutrophils were added in the top compartment and N-formyl-methionyl-leucyl-phenylalanine (fMLP) (10−7 M) was added in the bottom chamber. (D, E) Human neutrophils were stimulated in vitro with tumour necrosis factor α (TNFα) (100 ng/ml) or medium alone for 2 h and were assayed for qPCR analysis of IL-33R mRNA expression (D) or placed in the top and IL-33 (100 ng/ml) in the bottom compartment of the Boyden chamber (E). (F) Human neutrophils pretreated with TNFα or medium alone were placed in the top compartment and fMLP (10−7 M) was added in the bottom compartment of the Boyden chamber. Data are mean±SEM, human samples were evaluated individually, n=4–9. *p<0.05 vs medium alone (panels A and B), IL-33 top of Boyden chamber (panel A) or human healthy donors and anti-TNFα-treated patients with rheumatoid arthritis (panels B–F); †p<0.05 vs fourth column from right (panel A).
Discussion
The results of this study show that IL-33 is a potent chemo-attractant for neutrophils in AIA. It is principally produced by fibroblast-like synoviocytes and macrophages following an adaptive immune response. The precise nature of the induction of IL-33 synthesis by these cells is currently unknown but probably involves proinflammatory cytokines produced by T cells and macrophages following specific antigen challenge. IL-33 thus produced could effectively recruit the influx of neutrophils to the site of antigen challenge in at least two ways (figure 5): (1) it could trigger activated synoviocytes and macrophages to produce TNFα, IL-1β, CXCL1 and CCL3 which act in a cascade manner to recruit neutrophils to the joint; or (2) it could directly attract neutrophils to the site of inflammation. In further support of both mechanisms, fibroblast-like synoviocytes but not peripheral blood mononuclear cells of patients with RA produce IL-33 upon stimulation, and the IL-33 levels are higher in the synovial fluid than in the serum of patients with RA when measured simultaneously.38

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the proposed mechanism for interleukin 33 (IL-33) orchestration of neutrophil migration. (1) IL-33 is produced by synoviocytes and macrophage-like cells in response to antigen stimulation. (2, 3) IL-33 acts in an autocrine manner inducing cytokine and chemokine production. (4) Cytokines and chemokines induce endothelial cell activation and chemotaxis of neutrophils. (5, 6) IL-33 can directly attract neutrophils of antigen-primed mice or patients with rheumatoid arthritis by tumour necrosis factor α (TNFα)-induced IL-33R expression in neutrophils. (7) Neutrophils contribute to joint destruction.19,–,22 Steps 1–7 refer to the results in mice and steps 5–7 refer to the results in humans.
In the indirect pathway, activated synoviocytes and macro-phages appear to be the main producers and targets of IL-33 in an autocrine loop. IL-33 potently induces TNFα and IL-1 production12 which in turn are able to enhance IL-33 expression in synoviocytes.39 TNFα appears to act downstream of IL-33 but upstream of IL-1 in attracting neutrophil migration. IL-1, CCL3 and CXCL1 are all critically involved in the IL-33-induced neutrophil migration pathway and appear to induce the synthesis of each other. The cascade of cytokine/chemokine events induced by IL-33 following specific antigen challenge is reminiscent of the mechanism of action of IL-15 and IL-18 in the CIA and ovalbumin challenge in murine models of inflammation and neutrophil migration.29 32 However, IL-33-induced neutrophil migration is independent of IL-15 or IL-18 since it was unaltered by soluble IL-15 receptor α chain treatment or in IL-18–/– mice (see figure S2 in the online supplement). It is likely that IL-33, IL-15 and IL-18 act as parallel pathways, triggering a similar mechanism.
In an earlier report we showed that IL-33 exacerbates CIA in mice, at least in part, by activating mast cells.39 The finding reported here that IL-33-mediated neutrophil influx into the inflammatory site was mast cell-independent is therefore counterintuitive. It is noteworthy that the former39 and present studies were performed in different models of arthritis which could explain the mechanistic difference. It is likely that IL-33 enhances arthritis via several independent and interdependent pathways. IL-33 also chemoattracts human Th2 cells and activates eosinophils, basophils, mast cells, NK and invariant NKT cells.2 13 40 It is likely that, depending on the cytokine milieu, IL-33 could stimulate different cell types and assume different roles. The relative importance of these different IL-33-activated cells could vary depending on the disease context.
Besides participating in the cytokine/chemokine cascade for the induction of neutrophil migration, TNFα is also directly involved in the induction of IL-33R expression on neutrophils and hence priming neutrophils for a direct action of IL-33 on neutrophils. Bone marrow-derived neutrophils from naïve mice express little IL-33R and do not respond to IL-33-induced chemotaxis. In contrast, bone marrow neutrophils from antigen-primed mice or naïve neutrophils cultured with TNFα expressed substantial levels of IL-33R and responded readily to IL-33-induced migration in vitro. Moreover, TNFR1 deficiency abolished the chemoattraction by IL-33 of neutrophils from immunised mice concomitant with reduced IL-33R expression. This finding led us to investigate a potential novel mechanism for successful TNF targeting in RA.
IL-33R message was markedly increased in neutrophils from patients with RA treated with MTX alone compared with neutrophils from healthy donors. In contrast, IL-33R message was barely detectable in the neutrophils from patients with RA treated with MTX plus anti-TNFα (infliximab), a current standard treatment for RA. Moreover, only neutrophils from MTX-treated patients were chemoattracted by IL-33. Furthermore, after culturing with TNFα, neutrophils from healthy donors expressed as much IL-33R as neutrophils ex vivo from MTX-treated patients with RA and these cells then responded to IL-33 in chemotaxis. These results therefore support the clinical relevance of IL-33 in RA and provide an important novel mechanism by which anti-TNFα therapy ameliorates inflammation—that is, via inhibition of IL-33R expression in the neutrophils which inhibits neutrophil migration to the site of inflammation in response to IL-33. Thus, targeting IL-33 may represent a novel strategy against a range of inflammatory diseases associated with persistent neutrophil accumulation.
Acknowledgments
The authors thank Giuliana Francisco, Ieda Schivo, Sérgio Rosa, Cristiane Milanezi and Tadeu Vieira for their technical assistance.
References
Supplementary materials
Web Only Data ard.2009.122655
Files in this Data Supplement:
Footnotes
-
Funding This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo, Conselho Nacional de Desenvolvimento Científico e Tecnológico and Cordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil; the Wellcome Trust and the Medical Research Council, UK.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The Human Ethics Committee of the Faculty of Medicine of Ribeirao Preto (FMRP), University of Sao Paulo and the Ethics Committee on Animal Care and Handling Procedures of the FMRP approved this study.
-
Provenance and peer review Not commissioned; externally peer reviewed.