Article Text

Abstract
The understanding of the pathogenesis and optimal therapeutics for rheumatoid arthritis (RA) has advanced remarkably over the last decade. This review highlights these key advances, particularly the outcomes of genome-wide scans which have provided an increasingly robust appraisal of the complex genetics that underpin RA. Such observations are placed in pathogenetic context, particularly concerning the breach of tolerance that presages synovitis and the mechanisms that subserve chronicity. The key therapeutic strategies and treatment agents, both conventional and biological, now available to effectively manage the disease are described. Throughout the review, emphasis is placed on unanswered questions and challenges in this exciting field.
Statistics from Altmetric.com
Rheumatoid arthritis (RA) is a chronic inflammatory arthropathy associated with articular damage, attendant comorbidities, particularly in the cardiovascular system, and with increasing disability and socioeconomic decline. Such a pessimistic introductory remark may soon be rendered inappropriate given recent advances in the understanding and management of RA. Sophisticated genetic, molecular and cellular technologies applied via innovative translational discovery methodologies have elucidated the pathogenesis of RA to an unprecedented level. Novel therapeutics arising from such discoveries, coupled with revised and rationale-driven therapeutic strategies, have transformed the experience and prognosis for a substantial proportion of patients with RA. That said, significant unmet clinical need remains reflected in partial responses and the relatively low numbers of patients achieving sustained remission. In this review we summarise the recent key pathogenetic and clinical advances and thereby set out the state-of-the-art and challenges for a new decade.
Pathogenesis of RA
Much is now understood of the pathogenesis of RA as it evolves over time. The onset of arthritis is preceded by a prearticular phase comprising evidence of breach of self-tolerance (ie, autoimmunity)—for example, presence of anti-citrullinated protein antibody (ACPA), rheumatoid factor (RF)1 and lipid dysregulation and cardiovascular comorbidity.2 It is unknown whether subclinical synovitis is present at this time or whether a formal transition event must occur leading to clinically evident articular inflammation. The molecular nature of this transition is unclear, but probably reflects immune permissive microvascular, neurological or biomechanical factors. Clinically evident disease manifests within the joint with synovitis and attendant cartilage and bone damage. Recent advances have occurred in genomic analyses and in intensive analysis of the synovial lesion itself using advanced molecular technologies.
Recent genome-wide scans and validation analyses across discrete populations clearly implicate the immune system in incident risk and progression and have provided novel risk and modifier loci that can operate within or across these phases. To established risk loci in the major histocompatibility complex (MHC) class II region (eg, DR0401, 0404) has been added a variety of candidate genes that exhibit plausible biological effects. Many point to regulation of adaptive immunity, including ptpn22 (protein tyrosine phosphatase, non-receptor type 22 which regulates lymphocyte activation), ctla4 and cd40 (both implicated in co-stimulation of T cells). Other loci suggest modulatory influences on wider immune regulatory potential—for example, stat4, tnfa20, c5 (which in general regulate leucocyte activation and inflammatory signalling). A crucial recent advance has been the identification of gene–environmental interactions between MHC class II and smoking upon disease risk.3 The mechanisms underlying these interactions, and particularly how oral/pulmonary exposure and local citrullination in turn promote a localising arthropathy, are unclear but are likely to be highly informative for future preventive therapeutics.
The next challenge is to properly capitalise on this plethora of knowledge to inform the understanding of the pathogenesis at a cellular and physiological level—the application of functional genomics. Learning from other disciplines, particularly cancer, we suggest that this will likely be facilitated by the advent of a systems biology-based approach combining bioinformatic, proteomic, transcriptomic and metabolomic methodologies with high fidelity clinical phenotyping and information systems. Genetic approaches have already delivered a novel molecular taxonomy for RA. Thus, ACPA-positive disease may be separated from seronegative disease and should be considered a distinct clinical syndrome4 with attendant prognostic and comorbid features. The therapeutic implication of this has not been clarified but is already manifest in the optimal application of B cell targeting therapeutics. Future studies will deliver higher predictive value in biomarker-led predictive algorithms for prognosis and for therapeutic or toxic responses.
The synovial lesion in RA contains a macrophage/fibroblast-rich lining layer overlying interstitial tissues containing a plethora of activated leucocytes including macrophages (M1 phenotype), dendritic cells (DCs), B cells, CD4/CD8 T cells, mast cells, NK and NKT cells. A sophisticated trafficking system supports this infiltrate consisting of proliferative vascularisation reflecting the relative abundance of proangiogenetic factors, which exceeds the machinery normally necessary for cellular egress via lymphatics. Despite this vascularisation, the synovial lesion is intensely hypoxic, the biological significance of which is poorly understood.5 Formerly, single cellular dominance models of the synovial lesion (eg, ‘T cell-driven’, ‘fibroblast-driven’) were useful in creating and testing hypotheses. These have largely given way to integrated models of chronic inflammation that explain perpetuation on the basis of numerous positive feedback loops and failed regulation involving many cell types. Components of both the adaptive and innate immune response are implicated in these models. A critical role is likely for host tissue cells, particularly synovial fibroblasts, chondrocytes and osteoclasts.
Several emerging themes in the biology of synovitis merit particular attention. The core structural and topographical requirements for an inflamed synovial lesion are now emerging, addressing a fundamental question—namely, why should and how does the synovium facilitate the creation of a lymphoid-like tissue? Thus, a pivotal role for cadherin-11 and β-catenin (molecules that function to regulate cellular adherence and orientation properties of synovial fibroblasts) in maintaining the structural organisation of the synovial membrane in development and, importantly, during inflammation onset has been elegantly shown in human tissue and gene-deficient mice; the latter are incapable of sustaining articular inflammation.6 Similarly, the organisation and functional importance of innate and adaptive immune cells in synovium is becoming clearer. Numerous innate immune sensing pathways are found in the joint—specifically, Toll-like receptors (TLRs), NOD-like receptors and molecular components of the inflammasome are expressed and facilitate sensing of tissue damage.7 8 Mice deficient in various TLRs exhibit blunted or absent articular inflammation and ex vivo studies using particularly human fibroblast-like synoviocytes, B cells and macrophages implicate TLR-dependent pathways in aberrant (auto)antigen presentation, cytokine and matrix metalloproteinase production.9,–,11 The therapeutic utility of targeting these pathways is awaited.
In parallel, interest in adaptive immunity is driven increasingly by a desire to address a fundamental question in the biology of RA—namely, why is immune tolerance broken? It remains unclear whether this occurs in the joint or elsewhere in immune competent tissues such as the bone marrow or lymph node. It has long been recognised that activated CD4 and CD8 T cell subsets, B cells, plasmablasts and plasma cells are abundant in synovium. Ectopic germinal centres are present in a substantial proportion of patients, associated with expression of LTβ, CCL13 and CCL21; recent evidence shows that such structures can support B cell maturation and class switching (activation-induced cytidine deaminase-dependent) and thereby promote autoantibody production.12 The long-term prognostic significance of these ectopic germinal centres is, however, disputed and remains subject to ongoing analysis. Intriguingly, their presence may predict better responses to tumour necrosis factor (TNF) blockade,13 suggesting that they may define discrete cellular processes in some patient subgroups. The cellular basis for autoantigen presentation is also unclear; the success of B cell therapeutics and, in particular, reduction of autoantibody titres suggests that B cell lineages may have a role. Recently, however, both myeloid and plasmacytoid DC subsets (mDC and pDC) have been identified in RA synovium where they exhibit an intermediate maturation phenotype (likely antigen presentation capable) and high levels of cytokine expression.14 Model systems suggest that such mDC are proinflammatory whereas pDC may have net immune regulatory function.15 16 Their precise role in breach of tolerance, particularly in comparison with B cells, remains unclear; elucidating this will be important since DC modulation is currently being tested in clinical trials aimed at re-establishing immune tolerance.17 Their biology may also provide further rationale for earlier use of therapeutic co-stimulatory blockade.
Synovial cytokine biology remains a rich source of pathogenetic and therapeutic utility.18 19 RA was historically considered a Th1 disease. Studies of very early RA challenge this notion, implicating a more fluid T cell phenotypic view in which Th2-type responses may predominate to promote autoantibody production early.20 Identification of the importance of Th17 cells in murine autoimmune models has led to therapeutic developments across the spectrum of arthropathies.21 In RA, interleukin (IL)-17A and its regulatory moieties (IL-1, IL-6, IL-21 and IL-23) are expressed in established synovitis but with uncertain pathogenetic significance; clinical trials are ongoing to formally elucidate the pivotal nature of this subset and its regulatory cytokine network. Recent studies have identified several other cytokines of potential interest; for example, IL-33 is a novel IL-1-like cytokine that acts to integrate synovial mast cell and T cell activation,22 BLyS and APRIL are critical B cell survival factors likely to play a role in sustaining autoreactivity in the B cell compartment23 and adipocytokines which provide an intriguing link between adipose tissue activation, metabolic syndrome and articular inflammation.24 A major challenge is now to define the functional hierarchy and regulatory networks within which cytokines operate and, in particular, at which checkpoints their functions coalesce to optimise inhibitory strategies. Moreover, there is now increasing interest in defining those factors that in turn drive cytokine release. Extracellular factors such as immune complexes, TLR agonists and cell membrane-dependent pathways are pivotal. However, novel levels of regulation are emerging, including those mediated by microRNA,25 lipid sensing nuclear receptors26 and protease sensing receptor systems such as PAR2.27 Increasingly moreover, the signal pathways that subserve cytokine effector function and indeed synoviocyte activation are subject to scrutiny. This is driven on the premise that such pathways will provide rational drug targets. It has proved difficult to choose kinase activities of adequate disease specificity and tractability; notable failures reside in the p38 programmes that have consistently failed to deliver sustained benefit. However, the preliminary success in clinical trials of janus kinase and Syk kinase inhibition has renewed interest in this approach.28 29 Intriguing early data also support the potential for nuclear factor-κB, other mitogen-activated protein kinases (MAPKs) and sphingosine kinase-dependent pathways. Together these data raise the possibility of a new generation of orally bioavailable ‘biologic-equivalent’ agents.
A final important area of pathogenetic advance resides in efforts to integrate neuroendocrine interactions and articular inflammation. Gender imbalance in autoimmunity has long been recognised. Molecular explanations for such phenomena are increasingly provided in studies of endocrine-mediated regulation in animal models of inflammation30 and, in particular, in studies linking the hypothalamic pituitary adrenal axis and cytokine expression. Similarly, neuroimmunological inter- actions appear important in the development of arthritis in model systems. The CNS is implicated normally in immune regulation and homeostasis. Several neurotransmitters are expressed in RA synovitis.31 Cytokines are rapidly upregulated in the hypothalamus during inflammation and may participate directly therein, in perpetuation of psychological comorbidity. In arthritis models there is evidence for disruption of normally regulatory neuroimmune interactions.32 The therapeutic utility of these pathways is emerging; vagal nerve manipulation or α7AChR agonism has recently been shown to ameliorate collagen-induced arthritis.33 Similarly, manipulation of CNS MAPK function can mediate profound effects on peripheral arthritis.34 This is, however, a nascent field with many uncertainties as to the complex interactions ongoing.
Therapeutic strategies
Although the foregoing pathological advances have undoubtedly driven progress in RA treatment, major improvement in outcomes began with the acceptance of methotrexate (MTX) as a safe and effective agent in the 1980s,35,–,40 and gained critical momentum with the realisation that disease-modifying antirheumatic drugs (DMARDs) could safely and effectively be used in combinations.41,–,44 Progress then accelerated remarkably from the late 1990s with the introduction of an ever-increasing array of biological agents.45,–,48 We contend, however, that the most important paradigm shifts have been in recognising the importance of early treatment and in the understanding that clinicians should treat to a target of remission or very low disease activity. Moreover, there is emerging evidence to support intensive therapeutic regimens contained within such approaches.49 50 Discussion of therapeutic strategies by necessity should not concern specific drugs but rather focus on overarching therapeutic principles. The TICORA study was the first to examine this question specifically and provided important information. Simply stated, if physicians treat to a target, patients do better. Using a clinical target of Disease Activity Score (DAS) <2.4, patients receiving intensive therapy reached a mean DAS of 1.4 compared with a mean DAS of 2.4 in the ‘usual’ therapy group. Importantly, patients receiving intensive therapy not only improved clinically but had significantly less radiographic progression (median change in Sharp score 0.5 vs 3.9). The TICORA findings were particularly impressive since only conventional therapy was used. Other trials, while not specifically designed to address this question, have shown similar effects. In the BeSt study51 the key design feature was that clinicians treat to a target (DAS <2.4) by therapeutic adjustment. While there were differences among groups early in the trial, arguably the most important finding of the BeSt study was that 79% of patients achieved target DAS scores <2.4 at 2 years regardless of group.52 Accepting that it is critical to treat to a goal, two important questions remain: what should the goal be and how quickly do we need to get there? Should the goal be based on a DAS, DAS28, Simplified Disease Activity Index (SDAI), Clinical Disease Activity Index (CDAI), Routine Assessment of Patient Index Data (RAPID), Patient Activity Scale II (PAS-II) or some other measure? Should the goal be remission or low disease activity? If we use these largely clinical measures, how will radiographic progression or functional decline be factored in? Is it important to get patients to this goal in 2 months or is 6 months acceptable? Because of the efficacy of our therapies, another strategy question arises—when and what to stop in a patient who is in remission? Currently, there are essentially no data to address this latter increasingly common scenario, which represents a particular challenge for the coming decade.
Ultimately, ideal therapeutic strategies will be individualised for each patient and be dictated by a series of parameters including (but not limited to) genetic, metabolomic and transcriptomic profiles, soluble inflammatory makers together with markers of articular damage. Such biomarker-driven approaches will allow therapeutic stratification—the best informed choice for each patient. Until the arrival of validated biomarker algorithms, strategies should be built around a target of low disease activity or remission and, until we know how rapidly it is necessary to control disease, it appears that strategies and dose escalation or switching should be built around what is known about the time to maximum benefit of a given therapy. Currently there are no data to indicate that treating to goals defined by imaging studies—for example, x-rays, ultrasound scans or MRIs—provides long-term benefit; this represents a further important priority for investigation.
Pharmacological approaches in RA
The therapeutic armamentarium available to treat RA has expanded considerably in recent years and now comprises synthetic and biological DMARDs together with analgesics, cyclooxygenase-2 inhibitors and non-steroidal anti-inflammatory drugs (figure 1). The precise choice and order of DMARD use in the initial treatment of RA remains an individualised decision between patient and physician; however, early commencement of a DMARD should be considered optimal. The most obvious question to address is whether combination therapy—and, in particular, combination therapy with a TNF inhibitor—should be used at the outset or whether it is sufficient to step up to these therapies only in those patients who demonstrate the clinical need for them. Currently available data are equivocal. Most major studies that have compared initial combination therapy with initial monotherapy have shown that the combination group looks best initially43 44 51 53,–,57; however, only a very few of these studies51 have included a step-up arm which is critical to assess clinical relevance. To simply compare therapy A with therapy A+B and then, when patients in the A+B group look better at 1 or 2 years, to conclude that everyone should get A+B is an oversimplification of questions that are important to clinicians. For example, in the PREMIER study57 representing a direct comparison trial of early therapy, ACR20 responses were higher for MTX than adalimumab. The converse was true for radiographic progression; adalimumab alone was better than MTX alone. So, what is more important: clinical outcomes (ACR20) or radiographic outcomes (Sharp score progression)? The combination group did best in both measures but, since no step-up arm was included, some patients probably received unnecessary combination therapy and therefore added expense and toxicity risk exposure. Other trials with a similar design were ASPIRE54 and COMET,58 neither of which included a step-up arm. The BeSt trial compared the step-up approach with initial combination therapy in an unblinded way. Not surprisingly, the patients given initial combination therapy fared better in the early analysis51 but, by 2 years, the four groups were identical with regard to DAS.52 The groups given combination therapy first, however, had less radiographic progression at 2 years (median change in Sharp score of combination groups vs step-up groups = 1 on a scale of 0–448); the long-term consequences of this need to be further elucidated. The TICORA study used step-up therapy exclusively and the patients in the group treated to target fared well. Radiographic progression did continue in a minority of patients with a median progression in the intensive therapy group of 1.4 points per year (scale 0–448). The TEAR trial compared initial combination therapy with either MTX+etanercept or MTX+ sulfasalazine+hydroxychloroquine (triple, with step up from MTX alone to both combinations only if DAS28 >3.2.59 ] DAS28 scores were similar across the groups at 2 years with approximately 30% of patients maintained on MTX only. The TEAR trial therefore supports a step-up strategy. A similar comparison of the TICORA trial and initial triple therapy also provides no evidence for superiority of early combination approaches.60
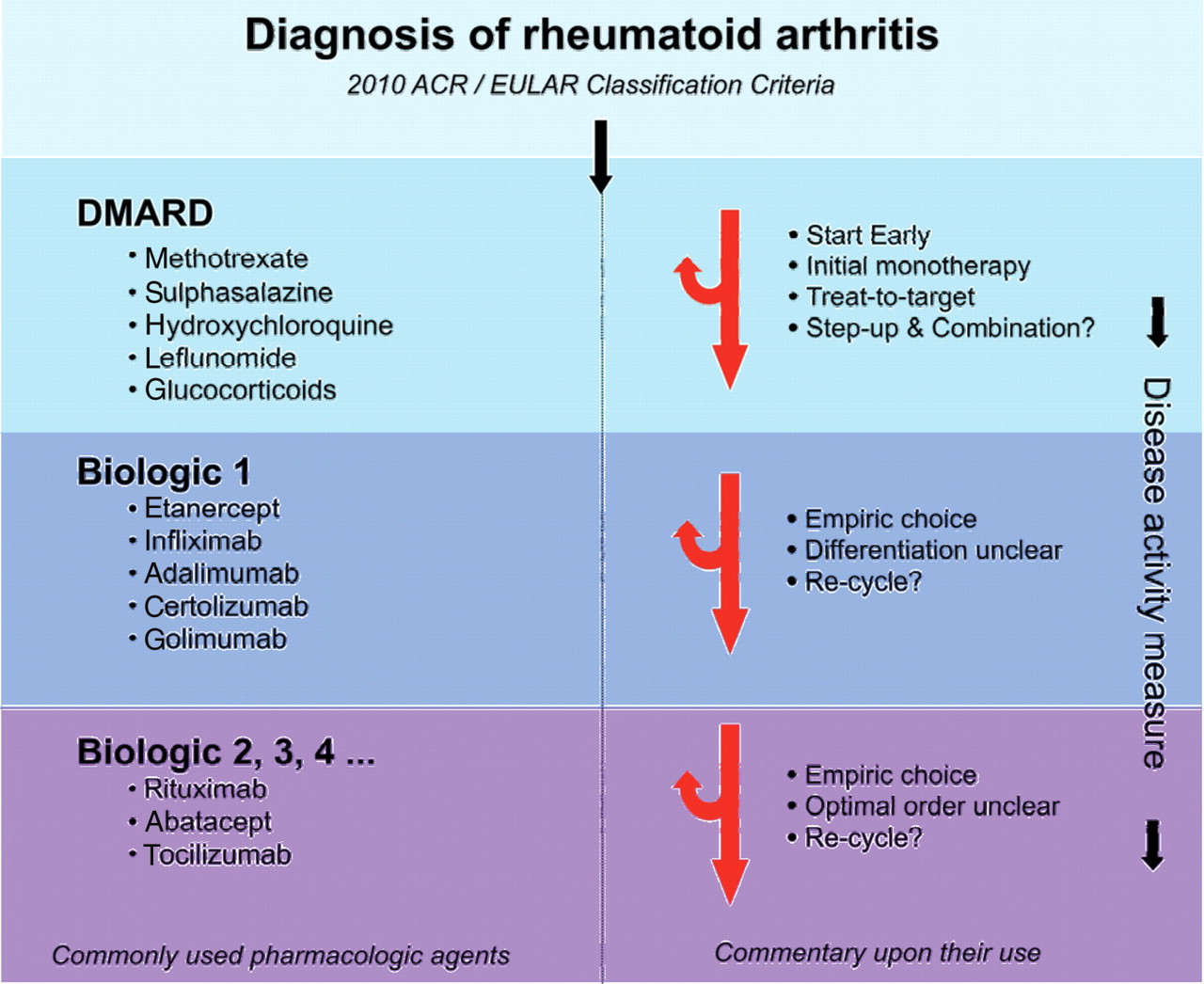
{kind=link}
Current treatment paradigm for rheumatoid arthritis (RA). The current management of RA is depicted to emphasise the sequential use of therapeutic agents driven by measurement of disease activity. The empirical nature of decision-making, despite advances in pathogenetic discovery, is highlighted. DMARD, disease-modifying antirheumatic drug.
Until other evidence emerges, therefore, initial therapy in the clinic should begin with monotherapy.1 However, clinical judgement should be used and, at the outset, TNF inhibition may rarely be appropriate in a subset of patients. The latter, however, is predicated on the notion that even a short delay (3 months) in biological therapy has a substantial future impact; data are required to prove this. MTX is not only the best studied DMARD but it is an important anchor drug of most successful combinations; all available data suggest that TNF inhibitors work best when used with MTX.56 57 MTX should therefore be the drug of choice unless there are contraindications, in which case sulfasalazine or leflunamide are useful options. The question of whether glucocorticoids should be added as part of initial therapy remains controversial, although recent guidelines conclude that their use is advantageous by oral or parenteral routes.61 62 Nevertheless, it is clear that glucocorticoids should subsequently be tapered rapidly if possible.
Patients with active disease despite MTX
When patients have active disease despite an adequate trial of MTX for 3 months, another agent(s) should be added. Trial data exist to support the use of many commercially available medications added to MTX including leflunomide,63 etanercept,46 sulfasalazine,64 hydroxychloroquine,64 sulfasalazine+hydroxychloroquine,64 infliximab,47 anakinra,65 adalimumab,66 gold,67 rituximab,68 abatacept,69 golimumab,70 certolizumab71 and tocilizumab.72 Only two published blinded trials have compared active therapies with each other in this group of patients: the first suggested that sulfasalazine+hydroxychloroquine was better than either alone,64 while the second showed similar efficacy for abatacept and infliximab.73 A third study was an open-label study which suggested that infliximab was better than sulfasala-zine+hydroxychloroquine at 12 months but not at 6 months.74 Recently published European League Against Rheumatism (EULAR) recommendations recommend a TNF inhibitor for this group of patients, particularly for those with poor prognostic factors (including early erosion, RF+, ACPA+ (in high titre) or high disease activity measures).62 This recommendation was based on a systematic review of the literature.75 We have highlighted above, however, that few trials have adequately addressed this critical question and suggest that this remains a contentious issue worthy of ongoing evaluation in formal trial designs. It will be important to properly understand the value of combination synthetic DMARDs in this patient group, particularly as health economic issues become more prominent. Trials designed in this group of patients that use placebo controls provide little useful information to clinicians and, because so many effective therapies are available, are probably unethical. Finally, based on the only study to compare biological agents in this group of patients,73 abatacept may also be considered; however, further analyses are required to address the optimal order of use of biological agents (see below).
Patients with inadequate response to combination therapy (including a TNF inhibitor)
Several approaches have been studied in patients who have failed the combination of MTX and at least one TNF inhibitor: switching to another TNF inhibitor,76,–,79 adding rituximab40 to existing MTX, adding abatacept to existing MTX80 or adding tocilizumab to MTX.81 The latter three approaches have been studied in randomised double-blind trials while switching to other TNF inhibitors (with the exception of golimumab) has been examined mainly in observational studies. No data comparing active therapies with each other are available in this group of patients. The trials that have added rituximab, abatacept, tocilizumab or golimumab to existing MTX have shown similar results.79,–,82 ACR20 responses have been seen in 35–50% of patients and reported severe adverse events have been similar. Observational studies have shown that switching from one TNF inhibitor to another is more likely to be successful if the reason for failure of the first TNF inhibitor was toxicity (ACR20 responses of ∼40%) than if the reason was true lack of or loss of efficacy (ACR 20–25%).
Conventional DMARDs and their current role
As discussed above, MTX remains the cornerstone of initial therapy and a key component of most effective combinations.35 We are still learning about its optimal use. The addition of low-dose folic acid supplementation has been standard practice for over a decade40 83 and is critical, at least in some patients, to decrease toxicities. Recent studies have again demonstrated issues with absorption of oral MTX in some patients, highlighting the need to split the dose into two on the day of administration if efficacy is inadequate84 or to switch to subcutaneous administration. Furthermore, recent work has shown that MTX does not reach steady state (at least in terms of red blood cell polyglutamation) until approximately 6 months of administration85 and, in most clinical trials, benefit continues to increase until 6 months of therapy. MTX should be rapidly escalated to at least 20 mg per week and given a minimum of 3 months to work. With this approach, approximately 30% of patients will achieve a DAS28 of <3.2 after 3–6 months.58 59 Folic acid should in most cases be given concomitantly. Hydroxychloroquine, sulfasalazine and leflunomide remain important (albeit often underused) components of the armamentarium. In general, these conventional DMARDs are used in combination with other DMARDs, frequently MTX. All have been shown to be effective in RA when used as monotherapy and when used in combination with MTX.41 44 50 51 With the increased use of biological agents, many clinicians now bypass these effective and relatively inexpensive DMARDs, although their role in some patients is retained in the current EULAR guidelines. The apparent decreasing use of hydroxychloroquine is perplexing given its capacity to reduce lipid metabolic risk and diabetes,86 both of which are likely to be important in patients with RA with attendant cardiovascular morbidity.
Biological therapy
TNF inhibitors
TNF inhibitors have been approved for clinical use for a decade and have changed forever the landscape of RA therapeutics. Currently, five TNF inhibitors are available. Four are monoclonal antibodies (infliximab, adalimumab, golimumab and certolizumab (PEGylated)) while one (etanercept) is a soluble receptor fusion protein. Efficacy in RA is remarkably similar among these products across many trials, often summarised as the ‘60–40–20’ rule. When using the ACR20, 50 and 70, one can expect 60–70% of patients to achieve an ACR20, 40% to achieve an ACR50 and 20% to achieve an ACR20, regardless of the preparation used.47 66 70 79 87 Similarly, all have been shown to substantially retard or abrogate radiographic progression. A variety of toxicities are now recognised with reactivation of latent tuberculosis,88 other granulomatous infections,89,–,91 soft tissue and joint infections,92 concerns for lymphoma,93 solid tumours90 93 and skin cancers, possible exacerbations of congestive heart failure, multiple sclerosis94 and new onset psoriasis95 worthy of mention. While toxicities can be devastating for an individual patient, most are rare and the majority of patients tolerate TNF inhibition well. Risk/benefit trade-offs need to continue to drive decisions and ‘numbers needed to harm’ should be derived to facilitate informed discussion with patients.93
A critical question is whether there are important clinical differences between the available TNF agents. While the similarities of both efficacy and toxicity are much more striking than differences with time, important differences are emerging. Monoclonal antibodies have clear advantages in treating inflammatory bowel disease and probably in treating inflammatory eye disease in RA and other conditions.96 97 They appear to be more likely to be associated with reactivation of tuberculosis88 and granulomatous diseases such as histoplasmosis89 and coccidioidomycosis as well as listeria infections.98 Additionally, observational data suggest that varicella zoster is more common with infliximab and possibly adalimumab than with etanercept.99,–,101 However, there is less evidence of any significant difference of the much more common problem of bacterial infection at the recommended clinical doses of infliximab, etanercept and adalimumab.91 92 On the other hand, although demyelination has been attributed to all of the TNF inhibitors, some data suggest that it may be more frequent with etanercept than with infliximab.94 As data accumulate, clinicians will be able to better select a treatment to match the clinical situation of an individual patient.
Biological agents after TNF blockade
Several biological agents are now available and are generally employed after TNF blockade. The long-standing association of RA with RF and the recent recognition of the importance of ACPA renders it surprising that B cell depletion was a relatively late development. Rituximab, a monoclonal antibody directed against CD20, is now approved and widely used in practice. It is unequivocally effective in RA,68 although its mechanism of action is not clear; this may include reduced autoantigen presentation, cytokine expression or autoantibody production. A single treatment course (usually two intravenous infusions) results in profound and long-lasting decreases in B cell (CD19) numbers. To maintain efficacy, retreatment is usually necessary some time between 6 and 12 months thereafter. While initial reports suggested that there was not a close correlation between B cell depletion and clinical response, more recent reports using more sensitive techniques to measure B cells have demonstrated a closer relationship; synovial biopsy studies indicate that plasmablast expression may be of particular importance. The clinical response appears to be significantly more likely in RF+ and ACPA+ patients, providing a useful biomarker for patient stratification.102 103 Since patients do develop prolonged and often profound B cell depletion, there are concerns about the long-term consequences of this and about the appropriate timing of retreatment or treatment with another biological agent after rituximab. Studies have shown significant reductions in immunoglobulin levels and possibly selective suppression of autoantibodies after treatment. A single study has reported on a series of patients given a second biological agent after rituximab without obvious added toxicity.104 Long-term toxicity remains a matter for careful registry follow-up, although early indications are encouraging. The impact on first-line biological use of rituximab regarding concern about the development of progressive multifocal leukoencephalopathy is currently unknown.
Abatacept (CTLA4-Ig) is approved and increasingly employed in the treatment of RA, particularly after TNF blockade.73 This phase III programme demonstrated efficacy in patients with an inadequate response to MTX and after TNF blockade failure, and thus far has exhibited a satisfactory safety profile. Recent studies in early disease show an intriguing increase in the level of efficacy achieved; whether this reflects an optimal mode of action in earlier pathogenetic stages or simply the often observed increased tractability of early disease to many agents is unclear. The mechanism of action of abatacept is poorly understood but may reside in the promotion of T cell anergy by prevention of the normal two signal activation requirement for T cells; TCR ligation in the absence of a CD28 signal promotes a subsequent hyporesponsive state in the T cell. Direct effects on follicular Th cells have recently been demonstrated.105 This raises the intriguing possibility in future of promoting T cell tolerance if used at an early or even undifferentiated phase of disease; formal studies will be required to confirm this possibility.
The most recent addition is tocilizumab, a monoclonal antibody that binds the IL-6 receptor and thereby inhibits the effector biology of IL-6. This exhibits impressive efficacy in patients with an inadequate response to MTX72 and similar responses in TNF blockade failure groups as were noted with rituximab and abatacept.106 So far toxicity appears reassuring; infections and rarely gastrointestinal perforation have been noted in the development programme. Significant alterations in plasma lipid concentrations have been observed in a variety of patients but, so far, this does not appear to be related to increased vascular pathology although long-term studies will be helpful in determining the magnitude of risk, if any. In the short term, proof of concept studies are ongoing that will offer mechanistic explanation.
The critical challenge that we now face is to properly allocate and order the use of biological therapeutics. In particular, beyond the safety reassurance of registries for longer licensed agents, it seems inappropriate in the medium term for order of discovery to be the deciding factor on order of use. Put simply, is there a rational mode of action for distinct phases of RA? Are there subsets of patients in whom one monoclonal antibody is superior or likely to be ineffective?
Biomarkers to drive decisions in therapy
The Holy Grail of RA is therefore prediction of natural history and, in therapeutics, of efficacy and toxicity. Significant progress has been made in understanding factors that predict poor prognosis including, for example, the presence and number of copies of shared epitope,101 ACPA or RF positivity,107 108 the level of acute phase reactants (erythrocyte sedimentation rate and C reactive protein) and erosions that are present at baseline. However, better understanding of prognosis does not by itself translate into improved therapeutic strategies. It is well-accepted that shared epitope-positive patients have a worse prognosis in general,109 and some data from several sources suggest that these patients respond less well to MTX,101 110 but this has not been found in all studies. RF and ACPA status may be useful in predicting the response to rituximab102 103; if a patient is negative for both, the chance of a favourable response is reduced. This may prompt use of therapies other than rituximab in the seronegative population although, ironically, fewer clinical datasets exist for newer biological agents in this subpopulation. A number of genetic factors or synovial biopsy phenotype have been reported in small trials to be associated with outcomes in anti-TNF-treated patients. None of these observations to date has been strong enough to differentiate the response sufficiently well to be clinically helpful and trials where treatment strategies have been based on these findings have not been published. The place of imaging in this area similarly remains unclear. A major challenge in the next decade will be to derive effective biomarker-driven algorithmic therapeutics.
Clinically useful outcome measures
If we are centred on treating to a target or goal,50 what should the goal be and how best should it be measured? To be useful an instrument must quantitatively measure absolute levels of disease activity. The ACR20111 or ACR hybrid112 that measure percentage of improvement, while very useful in clinical trials, are therefore not appropriate in practice. Most published trials have therefore used a variant of the DAS44 or DAS28.113 114 The need for a laboratory variable and the lack of functional or radiographic components may, however, limit the general utility of this in future. Several clinical instruments have been proposed that are simpler but still have excellent correlations with DAS; they include the SDAI,115 CDAI116 and several versions of the RAPID,117 PAS-II,118 Lundex and Mean Overall Index for Rheumatoid Arthritis (MOI-RA).119 120 All of these measures have the advantage of being easy to calculate using simple arithmetic as opposed to square roots and natural logarithms required for the DAS. Compared with the SDAI, the CDAI has the advantage of not requiring laboratory data so the results are available in the clinic in real time. RAPID and PAS-II have these advantages and incorporate a Multidimensional Health Assessment Questionnaire (MDHAQ) or HAQ-II and, importantly, RAPID3 and 4 and the PAS-II derive all data from the patient. RAPID5 incorporates a physician global score. For current practice it is more important that clinicians use and treat to a goal rather than which goal they use. Questions remain: regardless of whether a DAS, DAS28, SDAI, CDAI, RAPID, PAS-II or other measure are used, should the goal be remission or low disease activity? If we use these largely clinical measures, how to incorporate radiographic progression or functional decline?
Conclusions and challenges
The foregoing provides an encouraging picture of emerging pathogenetic understanding and a plethora of therapeutic options. Recent advances in clinical definitions embodied in the ACR/EULAR classification criteria for rheumatoid arthritis121,–,123 will provide an enhanced clinical basis upon which to build early intervention and pathogenesis discovery programmes. Future therapeutics and, in particular, the advent of orally bioavailable ‘biologic-similar’ agents will further enhance this portfolio. The optimal therapeutic strategy must be found and will be predicated on advances in clinical practice models, biomarkers and upon more sophisticated use of imaging modalities. In the meantime, clinical guidelines have been generated to optimise practice. EULAR has recently presented recommendations for the management of RA that provide practical advice on the optimal approach to patients as their disease progresses.62 Similarly, the crucial importance of treating to clinical disease activity targets has been embodied in the ‘treat to target’ initiative.124 Increasingly, measures of health utility and clinical effectiveness will be gathered and applied at earlier stages in new drug development in the application of novel therapeutic strategies. Earlier recognition of disease with a poor prognosis will facilitate patient stratification and improved long-term outcomes for the majority. Throughout this review we have highlighted challenges and questions for the next decade. Perhaps the most exciting issue, however, is the recognition of patients before the onset of arthritis; the real challenge resides in pre-arthritis recognition and the ultimate goal—RA prevention!
References
Footnotes
See Editorial, p 1895
-
Funding NIH, Wellcome Trust, MRC (UK) and Arthritis Research UK.
-
Competing interests IMcI has received grants or honoraria from Roche, Pfizer, Schering Plough and BMS who manufacture biologic agents used in the treatment of rheumatoid arthritis.
-
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- Miscellaneous