
Key Points
-
There is a misconception that targeted cancer therapies (TCT) incite fewer and less-severe toxicities than conventional chemotherapy; receptor crossreactivity between malignant and healthy cells can lead to off-target toxicities
-
Physicians should be aware of undesirable effects of TCTs — including cardiovascular, gastrointestinal, dermatological, and haematological — to help prevent or mitigate these effects, especially when they might reflect good treatment response
-
TCTs are generally considered to be better tolerated than conventional chemotherapies; however, economic costs associated with toxicities and symptom burden must be better clarified
-
More patients are choosing to continue TCTs late in the disease course, sometimes without clear clinical benefit, which poses new challenges for appropriate care at the end of life
Abstract
Molecularly targeted cancer therapies, such as small-molecule kinase inhibitors and monoclonal antibodies, constitute a rapidly growing and an important part of the oncology armamentarium. Unlike conventional (cytotoxic) chemotherapeutics, targeted therapies were designed to disrupt cancer cell pathogenesis at specific biological points essential for the development and progression of the tumour. These agents were developed to disrupt specific targets with the aim of minimizing treatment burden compared with conventional chemotherapy. Nevertheless the increasingly common use of targeted therapies has revealed some unanticipated, often clinically significant toxic effects, as well as compromising effective palliative and end-of-life management approaches. Although patients and clinicians welcome improvements in cancer prognosis, these changes can also impact patient quality-of-life. Therefore, as demand for oncology expertise increases, physicians need to apprise themselves of targeted therapies and their clinical implications, including drug-specific side effects, impact on quality of life, and cost issues, especially in relation to end-of-life care. This Review provides a useful summary and guide for professionals treating patients with malignant diseases.
Similar content being viewed by others

Introduction
Targeted cancer therapy (TCT; also known as molecular targeted therapy or biological therapy) refers to a diverse group of agents that, unlike conventional (cytotoxic) chemotherapy, block the growth and progression of malignant cells by interfering with distinct pathways that are considered essential for the development and growth of various tumours1,2. These agents include small-molecule tyrosine kinase inhibitors and monoclonal antibodies (mAbs). Conventional chemotherapy interferes non-selectively with the proliferation of all rapidly dividing cells in the body, and therefore exerts cytotoxic effects on non-cancerous cells, such as haematopoietic cells of the bone marrow, epithelial cells of the gastrointestinal tract and cells of hair follicles. The rationale for the development of TCTs has been to specifically block one or more molecules that are either upregulated and overexpressed or mutated in tumour cells, thus minimizing toxicities, while improving treatment effectiveness. In the past decade, the development and use of TCTs has grown substantially3. Among these drugs, trastuzumab to treat HER2-positive breast cancer, and imatinib for the treatment of chronic myelogenous leukaemia (CML) and gastrointestinal stromal tumours, are considered among the greatest successes of TCT development, prolonging patient survival and decreasing disease recurrence4,5,6.
However, it became apparent that TCT actions do not restrict themselves to tumour cells, and TCT is not without adverse effects or complications7. While the available data on TCT-related toxicities in randomized clinical trials (RCTs) is limited owing to trial design, patient selection, reporting issues or other reasons, a meta-analysis of the safety and tolerability as reported in 38 RCTs assessing agents for the treatment of solid tumours that were approved by the US FDA between 2000 and 2010, found greater risk of death due to toxicity with TCTs (pooled odds ratio (OR) 1.4; 95% CI 1.15–1.7; P <0.001), treatment discontinuation (OR 1.33; 95% CI 1.22–1.45; P <0.001) and grade 3 or 4 adverse events (OR 1.52; 95% CI, 1.35–1.71; P <0.001) with TCTs than with control groups8. Importantly, statistical significance remained after placebo and best supportive care control groups were excluded as comparators. The compared trials were very heterogeneous and this analysis has limitations; nonetheless, it provides valuable quantitative information on the risks associated with TCT use, which were higher for non-haematological than for haematological toxicities. A more-recent systematic review and meta-analysis of 43 RCTs performed with VEGFR or EGFR-family targeted kinase inhibitors, despite being associated with significant improvement of progression-free survival (PFS, hazard ratio (HR) 0.82; 95% CI 0.76–0.89), but not overall survival (HR 0.99; 95% CI 0.95–1.03), confirmed that addition of kinase inhibitors to chemotherapy significantly increased the risk of fatal adverse events (relative risk (RR) 1.63; 95% CI 1.32–2.01), treatment discontinuation (RR 1.8; 95% CI 1.8–2.06) and any severe adverse event (RR 1.25; 95% CI 1.16–1.36)9. Although it can be argued that the combination of drug classes increases the likelihood for toxic effects, combination therapies are not unusual in the treatment of malignancies; in fact combination therapies are required in many cases because single-agent targeted therapy lacks sufficient efficacy for several indications. Approximately two thirds of cancer incidences as well as mortalities occur in patients 65 years or older, who are burdened with various comorbidities that are not related to their malignant disease. Such comorbidities include hypertension, cardiopulmonary diseases, diabetes, impairments of hepatic or renal functions, or other conditions that may require medical treatment10. Interactions with those medications can affect the pharmacokinetics of many TCTs11.
At this time, TCT dosing is commonly based on efficacy parameters determined in clinical trials, but also considers a patient's ability to tolerate potential side effects or even more serious adverse events. As TCTs have an expanded role in the treatment of malignant diseases, awareness of the undesired symptoms they may cause are key. This Review provides an overview of the development and toxicity profiles of the two main types of TCTs, kinase inhibitors and mAbs, with an emphasis on their clinical management. Some observed toxicities differ from those seen with conventional chemotherapies, while others are similar or overlapping, such as cytopenias, gastrointestinal toxicity, fatigue or thromboembolic events. Because of the paucity of available data this Review has not addressed the implications of potential drug–drug interactions for patients and the role they have in the clinical outcomes. We also raise awareness of the needs of patients with cancer for supportive care and timely referral to hospice services, as the use of TCTs may delay such referrals, and we discuss the impact of TCT use on the health-care expenditures at the end of life.
Modes of actions of TCTs
Kinase inhibitors are small-molecule drugs that are are derivatives of pyridines (crizotinib, sorafenib), pyrimidines (dasatinib, imatinib, nilotinib, pazopanib), quinolones (bosutinib), quinazolines (afatinib, erlotinib, gefitinib, lapatinib, vandetanib) or carboxamides (dasatinib, regorafenib, sunitinib), and these agents exert their effect intracellularly. Orally administered, kinase inhibitors competitively inhibit adenosine triphosphate at the catalytic binding site of the targeted intracellular enzyme, directly compete with the substrate of one or more-specific kinases, or bind at alternative sites to induce conformational molecular changes that inhibit enzyme activity12. The toxicity profiles of kinase inhibitors depend on which kinase was targeted and on the role the enzyme plays in intracellular signalling and overall cell function.
mAbs differ from kinase inhibitors in their modes of action at the target level13,14. These agents are too large to penetrate cell membranes, so mAbs are designed to bind selectively and specifically to epitopes expressed on the surface of cancer cells. mAbs have a longer half-life than kinase inhibitors and are, in general, administered intravenously. They can either interfere with signal transduction from the cell surface, act in a stimulatory fashion to induce cellular events, such as apoptosis, or provoke an immune response against the targeted cancer cells possibly interferring with proteins that are necessary for the growth of cancer cells13. mAbs can be used as single agents, or combined with chemotherapy or hormonal therapy. The antitumour effect of 'naked' mAbs can be enhanced by linking them to cytotoxic drugs15, radioisotopes, certain cytokines or other agents (that is, conjugated mAbs). The first mAbs were nonhuman and generated by hybridoma technology, where myeloma cells were fused with spleen cells of mice that had been immunized with a desired antigen16. The nonhuman nature of the early proteins, and their repeated administration was hampered by inflammatory reactions and the development of human anti-murine antibodies in recipients. Consequently, recombinant technologies were used to engineer antibodies in viruses or yeast. With these technologies antibodies with partially human sequences, and thus higher degree of similarity to natural human antibodies could be generated. In chimeric mAbs (human immunoglobulin constant domain and murine variable domain) up to two thirds of the murine DNA is replaced by human DNA, whereas in humanized mAbs (murine complementary-determining region introduced into human immunoglobulin G) the proportion of human DNA is increased even further to up to 90%17,18. Fully humanized mAbs, such as adalimumab and panitumumab, do not contain any nonhuman components19. As these humanized mAbs have fewer foreign species sequences, not only have problems related to immunogenicity in recipients been considerably reduced, but the specificities of the mAbs for target epitopes have also been increased20,21,22,23.
Classifications of adverse effects
The diverse nature of TCTs and the heterogeneity of responses in different individuals pose a challenge to developing a classification based on patient clinical symptoms. TCT pharmacokinetics depends on inter-individual variations of drug absorption, distribution and metabolization, and is influenced by the pharmacogenetic background of a patient (such as cytochrome P450 and ABC drug transporter polymorphisms)24. In general, kinase inhibitors are less target-specific than mAbs25 and can penetrate the inside of cells, causing direct effects on cells, whereas mAbs have indirect effects and induce immune responses13. In addition, mAbs increase the likelihood of potential adverse immunological reactions13,14.
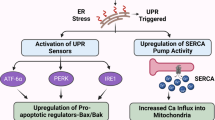
In 2005, Lee and Kavanaugh suggested distinguishing between target-related and agent-related adverse effects of biological agents26. In 2006, Pichler proposed to classify adverse effects of biologic agents based on their mechanism of action and structure27. In Pichler's classification system, type-α adverse effects are those related to release of cytokines, which can present mildly with flu-like symptoms, fever, chills, nausea, vomiting, hypotension, dyspnoea, etc., but can progress to a more-severe presentation associated with organ failure and even death. Type-β reactions are either immediate IgE-mediated or delayed IgG-mediated or T-cell mediated hypersensitivity reactions. IgE-mediated reactions can present as hives and/or pruritus, but also as acute urticaria and anaphylaxis, while IgG- and T-cell mediated reactions are characterized by myalgia, arthralgia and dysphagia. Type-γ adverse events are related to immune imbalance unrelated to high concentrations of cytokines or hypersensitivity. Type-δ adverse effects are due to antibodies originally directed against tumour antigens, but unintendedly cross-reading with antigens on non-cancerous cells, while type-ε adverse effects are essentially non-immunologic, such as heart failure, neuropsychiatric symptoms (acute confusion, depression) or retinopathies27.
Pretreatment and premedication assessment
Patients must undergo laboratory evaluation (complete blood count, urine analysis, pregnancy test, thyroid function test), and cardiovascular and pulmonary assessments before receiving kinase inhibitors or mAbs. When the drugs are given intravenously, patients are usually premedicated with acetaminophen, antihistamines, steroids and non-steroidal anti-inflammatory drugs (NSAIDs). If patients develop undesirable symptoms, management will depend on the degree and nature of those symptoms, and administration of the targeted agent might need to be discontinued temporarily or even permanently.
Kinase inhibitors
Common adverse effects of kinase inhibitors
There are at least 100 signalling pathways in humans28, and depending on the specific target inhibited, the adverse event profiles of kinase inhibitors can differ considerably, and can develop insidiously. Moreover, based on the oral administration of kinase inhibitors the measured plasma concentrations in pharmacokinetic studies and the bioavailabilities of the drugs can vary considerably between patients, even when given at standard doses24.
In general, most kinase inhibitors cause cytopenias; for example, in the prospective, multicentre, open-label, phase III IRIS RCT, imatinib was compared with IFNα and low-dose cytarabine for newly diagnosed CML29. Grades 3 or 4 anaemia, neutropenia and thrombocytopenia were reported at 3.1%, 14.3% and 7.8%, respectively, after a median follow-up of 19 months with imatinib versus 4.3%, 25% and 16.5% with the IFNα and cytaribine combination. Other kinase inhibitors can induce different degrees of cytopenia.
The digestive system is also affected by kinase inhibitors, which can cause nausea, vomiting, diarrhoea and/or heartburn7 (Table 1); some agents cause headaches, muscle cramps, periorbital oedema, and various types of skin rashes to varying severity levels30. Kinase inhibitors might induce or worsen symptoms of depression. They are teratogenic, and female patients of reproductive age should take appropriate measures to prevent pregnancy and/or stop breast-feeding during treatment31.
Black-box warnings have been issued for several kinase inhibitors. They are listed in Box 1. Kinase inhibitors can interact with a variety of drugs, such as H2-blockers or proton pump inhibitors, and with CYP3A inhibitors (such as atazanavir, clarithromycin, indinavir, itraconazole, ketoconazole, nelfinavir, ritonavir, saquinavir, voriconazole) or CYP3A inducers (for example, carbamazepine, dexamethasone, phenobarbital, phenytoin, rifampin, rifabutin, St. John's wort). A cross-sectional study conducted to assess the epidemiology of exposure to drugs with a potential to interact in patients receiving systemic anticancer agents, found potential interactions in 27% of patients, 9% of these interactions were classified as severe and 77% as moderate32. Approximately 87% of the interactions involved agents not related to cancer treatment (warfarin, antihypertensives, corticosteroids, anti-convulsants), while 13% were antineoplastic drugs, and 49% of the potential interactions were supported by level 1 or 2 evidence32. Moreover, the OR for increased risk of potential drug interactions was 1.4 per each drug added (95% CI 1.26–1.58; P <0.001)32. Although the study did not comment on the frequency of adverse clinical consequences and implications of the potential drug-drug interactions in patients, awareness of potential interactions and careful review of each patient's medication lists is advised. Consumption of grapefruit, grapefruit juice and Seville oranges during treatment can inhibit CYP3A4 activity on the intestinal wall and result in alterations of the bioavailability of kinase inhibitors. Resistance to kinase inhibitors may develop, possibly due to the existence of stem-like cells that protect cancers from drug effects33,34; such an outcome necessitates increasing the drug dose to achieve an effect, thereby increasing the risk of further adverse events, or ultimately a switch to a different agent.
Monoclonal antibodies
Nomenclature
Monoclonal antibodies that are derived entirely from murine gene sequences contain the syllabus –o (for example, tositumomab), while those that are chimeric contain the syllabus –xi (for example, rituximab). Humanized mAbs contain the syllabus –zu (for example, trastuzumab), and those that are fully humanized contain the syllabus –mu (for example, panitumumab).
Common side effects of mAbs
The most common adverse effects of mAbs are allergic reactions, such as infusion reactions, hives and/or pruritus, flu-like symptoms, including fatigue, chills, mild fever, headaches, muscle aches, gastrointestinal upset with nausea, vomiting and/or diarrhoea, hypotension, and skin rashes (Table 2). The black box warnings issued for mAbs are summarized in Box 2. Resistance to mAbs may be due to the combination of tumour-related and host-related factors, but complement-dependent and antibody-dependent cellular cytotoxicities are considered to be involved in the development of resistance mechanisms13,35.
Managing specific side effects of TCTs
There is a misconception that TCTs create fewer of the classic toxicities associated with conventional chemotherapies, such as diarrhoea, nausea, vomiting, alopecia or neutropenia, and that the toxicities experience with TCTs are less severe than conventional chemotherapy agents. As the targets of kinase inhibitors and mAbs are varied and widely distributed, off-target toxicities can develop that are related to receptor crossreactivity between the target in cancer cells and healthy cells in patient. In addition, on-target toxic effects related to drug mechanisms may be experienced30. While the former do not usually correlate with treatment efficacy, the latter have been linked to response to treatment, such as skin rash that develops with use of EGFR inhibitors36. The correct differentiation of on-target and off-target effects is needed for proper management.
Cardiovascular toxicities
Cardiotoxicity associated with TCTs28 seems to lead to rapid ventricular dysfunction, which is rarely observed with conventional cytotoxics. Ventricular dysfunction with TCTs can be less severe and has the potential for partial recovery37, either after discontinuation of the drug or with additional medical therapy. This outcome difference has been attributed to the different mechanism of cardiotoxicity of these drugs. While anthracyclines cause ultrastructural changes in cardiomyocytes38, thus directly affecting cardiomyocyte survival, cardiotoxicity associated with kinase inhibitors results from inhibition of ligand-related signalling pathways, as kinase inhibitors frequently interact with several ATP-binding sites37. Cardiotoxicities with kinase inhibitors manifest as a result of on-target as well as off-target effects, for example, as noted with imatinib, sunitinib and sorafenib39,40.
Chemotherapy-related left ventricular (LV) dysfunction causes congestive heart failure (CHF) in approximately 1–5% of patients; the range for asymptomatic decrease of LV function is 5–20%41. Among TCTs, LV dysfunction has been observed particularly in association with HER2 inhibition. The estimated absolute increase in risk for LV dysfunction with trastuzumab is 7.2–7.5% in patients with early-stage disease and 1.6–1.9% in late-stage breast cancer; it is generally lower with pertuzumab or lapatinib, but can be as high as 27% if a HER2 inhibitor is given in combination with anthracyclines42,43,44,45,46. Other kinase inhibitors that potentially induce LV dysfunction include anti-angiogenic agents (sunitinib, sorafenib, vandetanib), and inhibitors of MEK (trametinib) and ABL (imatinib, nilotinib, dazatinib) pathways30. Manifestations can range from asymptomatic electrocardiogram findings to severe CHF. Predisposing factors include advanced age, history of cardiac disease, previous anthracycline therapy and kinase inhibitor-induced hypertension47. The best management is prevention, with cardiovascular haemodynamics optimized before initiating therapy. Monitoring vital signs, cardiac function by laboratory assessment (troponin, pro-brain natriuretic protein), echocardiogram and multi-gated acquisition (MUGA) scan are indicated with some drugs, such as trastuzumab, and in symptomatic patients. Standard treatment of CHF with angiotensin converting enzyme inhibitors, beta-blockers, diuretics and/or aldosterone receptor blockers might be required, but beta-blockers are not tolerated well by some patients37. A decline of LV ejection fractions (LVEF) by more than 10 EF units has been reported in approximately 30% of patients receiving sunitinib or sorafenib48,49. A drop of LVEF below 40% might indicate withholding the targeted agent. LVEF should be re-assessed 4–6 weeks thereafter to determine the degree of recovery. The targeted agent can be resumed upon partial or complete recovery of the LVEF. However, balancing the risks versus the benefits of doing so must be individualized.
Hypertension is observed most frequently with VEGFR inhibitors, such as axitinib, lenvatinib, pazopanib, regorafenib, sorafenib, sunitinib, vandetanib, and bevacizumab30,50. This on-target toxicity usually occurs within the first month of treatment, but can occur even as early as within 24–48h upon treatment initiation. The overall incidence ranges between 20–30%, but the incidence rates can vary for individual agents50. For mild-to-moderate hypertension, observation alone can suffice. There are no specific guidelines regarding pharmacological intervention for severe TCT-induced hypertension, except that non-dihydropyridine calcium channel blockers (CCBs), such as diltiazem or verapamil that inhibit CYP3A4, should be avoided in conjunction with bosutinib, ibrutinib, pazopanib, sunitinib, and other TCTs as the interactions might result in dosage alterations of the targeted agent. Hypertension with VEGFR-inhibiting kinase inhibitors and mAbs, particularly bevacizumab, can cause posterior reversible encephalopathy syndrome (PRES)51,52.
Pulmonary arterial hypertension has been reported for dasatinib53. It is an off-target effect of the drug with an average incidence rate of 0.45–1.2% among treated patients54. It is dose-dependent and can develop after a latency period for up to 1 year. However, it is generally reversible with drug discontinuation. Change of cardiac repolarization — QTc interval prolongation on electrocardiogram — is an on-target as well as off-target effect that occurs with variable frequency50. Specific management guidelines are lacking, but the prolongations are rarely clinically worrisome. Caution is advised in patients with underlying cardiac disease and when drugs with known or possible risk of QTc prolongation are used, such as amiodarone, macrolide and quinolone antibacterials, 5-HT3 anti-emetics, and anti-retroviral protease inhibitors47. QTc prolongations to >500 ms with kinase inhibitors or >60 ms from baseline may increase the risk of triggering potentially life-threatening ventricular tachyarrhythmias, in particular TdP, which, when sustained, can convert to ventricular fibrillation and might be fatal. Therefore, repeated electrocardiograms are required while patients are treated with certain kinase inhibitors, particularly nilotinib, vandetanib or vemurafenib50. It is important to correct serum electrolyte imbalances, such as hypokalaemia or hypomagnesaemia, which represent additional risk factors that can facilitate and/or trigger cardiac arrhythmias.
Bradyarrhythmias do not typically occur with kinase inhibitors or mAbs, except with the ALK inhibitor crizotinib, where grades 1 and 2 bradycardias have been reported in 5% of treated patients55. Decrease in heart rate has been hypothesized to be associated with higher response rates to treatment56,57, but definitive conclusions cannot be made.
Thromboembolic events
Thromboembolic events are reported primarily for kinase inhibitors and mAbs that interfere with the VEGF pathway. Several studies have confirmed the association of bevacizumab with arterial thromboembolism (ATE)58,59,60, while the risk of venous thromboembolism (VTE) is not as well recognized60,61,62,63. The bevacizumab-related incidence rate of ATE is low at 2–3%, but it is twice as high as in patients treated with conventional chemotherapy64. A meta-analysis of 20 RCTs that assessed 13,026 patients found the relative risk for ATE events to be 1.46 with administration of bevacizumab compared to patients who were treated with placebo or non-bevacizumab-containing regimens65. Patients with renal cell cancers and colorectal carcinomas were at particularly high risk for ATE. Incidence was increased if patients used concomitant anti-thrombotic therapy with low-dose aspirin, although this was not statistically significant (5.1% in patients who received chemotherapy plus bevacizumab versus 1.2% in patients treated with chemotherapy alone; P = 0.159)58. The relative risk for ATE with sunitinib or sorafenib was 3.03 compared with controls (95% CI 1.11–1.93; P = 0.007) in a meta-analysis of 10 RCTs that assessed 10,255 patients66. The relative risk of VTE with VEGFR kinase inhibitors versus controls was evaluated in a recent meta-analysis that included eight phase II and nine phase III RCTs encompassing a total of 7,441 patients. The relative risk was 1.1 (95% CI 0.73–1.66; P = 0.64) with kinase inhibitors and was not statistically significant compared to 0.85 for the control arms (95% CI 0.58–1.25; P = 0.64)67. The risk increased with age above 65 years and a prior history of thromboembolism. Thus, it is currently not recommended to give aspirin prophylactically in this context to reduce thromboembolic event risk.
Haematological toxicities
Myelosuppression is the most frequently observed haematological toxicity of kinase inhibitors and mAbs. Management must be tailored to the cell type that is reduced in the peripheral blood. Neutropenias can put patients at risk for bacterial and fungal infections. Febrile neutropenia (FN) is not uncommon with conventional chemotherapy and the mortality due to this complication is approximately 2–4%; however, FN is far less frequently observed with TCTs, although neutropenia without fever is seen often with Bcr-Abl inhibitors. Dasatinib and imatinib were associated with grades 3 and 4 neutropenia in 21% and 20% of patients, respectively68. In the rare event that FN occurs with TCTs, treatment interruption may be required and full infectious disease evaluation is recommended, including obtaining blood and urine cultures, as well as assessment and possibly removal of any type of indwelling vascular device. Treatment is identical to the treatment of FN secondary to conventional chemotherapy. Early administration of broad-spectrum antibiotics may be indicated, particularly in high-risk patients, such as those older than 65 years, those with underlying comorbidities, or those with hepatic or renal dysfunction. Haematopoietic growth factors, such as granulocyte macrophage colony-stimulating factor (GM-CSF; sargramostim), granulocyte colony stimulating factor (G-CSF) (lenograstim and filgrastim) and pegylated filgrastim can be given to support faster recovery of the neutrophil count, although they do not reduce mortality rates, but reduce the length of FN-related hospitalizations69. Notably, in the event of concurrent corticosteroid administration, neutropenic patients may not develop fever, which is frequently the only symptom of an infection, but instead can be 'covertly' at risk for infections and/or sepsis. Therefore, special attention must be paid to these patients.
Mild anaemia and thrombocytopenia do not usually require treatment, but when more pronounced, transfusions of blood products are needed. Thrombocytopenia below 10,000–20,000 cells/μl increases risk of haemorrhage at the tumour sites or other sites of the body. In the event of a haemorrhage, patients may sometimes require prolonged hospitalizations and intensive care, which significantly increases the total cost of their treatment. Other manifestations of haematological toxicities might include bone marrow (BM) aplasia, BM necrosis, and gelatinous BM transformation with Bcr-Abl inhibitors, and thrombotic-thrombocytopenic purpura/haemolytic uraemic syndrome with VEGFR inhibitors, such as sunitinib70.
Gastrointestinal toxicities
Diarrhoea is a common adverse effect of conventional chemotherapy and experienced in 50–80% of patients depending on the regimen (≥30% grades 3–5 according to the common terminology criteria for adverse events corresponds to severe or medically significant, but not immediately life-threatening consequences (grade 3), life-threatening consequences requiring urgent intervention (grade 4) and consequences resulting in a patient's death (grade 5)71. It occurs primarily with (bolus) administrations of 5-fluorouracil (5-FU), with capetabine or irinotecan. Diarrhoea can occur immediately after drug administration or with a delay of at least 24 hours thereafter. The underlying pathophysiological mechanisms are multifactorial and vary between the drugs, as a result of mitotic arrest of crypt cells with 5-FU or owing to genetic polymorphisms of UGT1A1 and the presence of the active metabolite SN38 in the intestines as a result of irinotecan treatment71,72,73.
Diarrhoea occurs in approximately 60% of patients receiving TCTs; up to 10% are noted to be severe74. Kinase inhibitors are more likely than mAbs to induce diarrhoea, which can be dose-limiting but it can also indicate response to treatment, as reported for gefitinib75. Gefitinib-induced diarrhoea might be related to thromboxane A2-mediated changes, as a nonrandomized study led to symptom improvement with administration of low-dose aspirin76. Other mechanisms of diarrhoea might be related to drug dose, as noted for imatinib77,78 or to excessive chloride secretion and deficient sodium absorption79,80, which has been observed with EGFR inhibitors. Routine evaluation including screening for Clostridium difficile toxin and obtaining stool cultures is indicated. Diarrhoea management remains generally supportive with agents that decrease intestinal motility, such as loperamide81. Additional measures must be taken to prevent dehydration; depending on the type of targeted agent used and the indication, dose reduction, treatment interruption or even drug cessation and change of therapy may be required.
Nausea is another common adverse event of TCTs, and antiemetic agents can be considered, such as phenothiazines (prochlorperazine, promethazine), 5-HT3 serotonin antagonists (ondansetron, granisetron), dopamine (D2) receptor antagonists (metoclopramide), and benzodiazepines (lorazepam). Anti-psychotic agents (haloperidol, olanzapine) can be indicated in some cases, although no guidelines exist regarding specific pharmacological interventions.
Hepatic complications
Hepatotoxic manifestations of kinase inhibitors can range from mild transaminitis to severe drug-induced cytolytic hepatitis with necrosis, reported for imatinib82,83,84,85,86. Imatinib can also cause autoimmune-mediated hepatitis87, and hepatotoxicity has been reported to be one of the main causes for discontinuing imatinib treatment. However, despite several black box warnings regarding hepatotoxicity, clinically significant alterations of liver parameters are rare with kinase inhibitors, and are less likely noted with mAbs74,88,89. Management must be tailored to the underlying aetiology and be individualized to each patient90. To minimize complication risks, serum liver parameters should be assessed before initiating TCTs and repeatedly while on treatment with TCTs. In addition, it is advisable to avoid combinations with other hepatotoxic drugs. Once grade 3 toxicity is noted (defined as raised serum aminotransferase or alkaline phosphatase levels, total serum bilirubin level >2.5 mg/dl and hospitalization, or prolongation of pre-existing hospitalization because of drug-induced liver injury) discontinuation of the targeted agent, close monitoring of the patient and liver function by laboratory evaluation, and empiric treatment and supportive care represent the most reasonable management approaches. Some kinase inhibitors, such as erlotinib, nilotinib, pazopanib, regorafenib and sorafenib, can increase the unconjugated serum bilirubin fraction. If not severe, treatment can be continued.
Dermatologic toxicities
Dermatological adverse events remain a consistent, clinically significant phenomenon with TCT, albeit often poorly understood by clinicians30,91. These syptoms are frequently unpleasant and often reduce patient quality of life (QoL)92. These adverse effects develop in a dose-dependent fashion, particularly with EGFR inhibitors (erlotinib, gefitinib, cetuximab, panitumumab), VEGFR (sorafenib, sunitinib, pazopanib) and RAS/RAF/ERK (dabrafenib, vemurafenib) pathway inhibitors. The acneiform papulo-pustular skin rash seen with EGFR inhibitors is considered a surrogate marker of treatment efficacy and response36. In some cases, however, the severity of the rash may cause disfigurement and, thus, necessitates temporary treatment discontinuation or even permanent cessation. Available studies suggest that skin toxicities from EGFR mAbs, such as panitumumab, are more severe than those from kinase inhibitors; however, evidence-based prevention or management guidelines are not available. Male patients and patients younger than 70 years have been found to be at risk for developing rash related to EGFR mAbs93. Before therapy initiation, all patients are advised to avoid sun exposure, use sunscreens, enhance skin moisturizing, and avoid skin friction94. No detailed management guidelines exist for acneiform rash; experts recommend topical antibiotics such as clindamycin 1% +/− benzoyl peroxide, or oral antibiotics (tetracycline, minocycline or doxycycline)94, which may be prescribed prophylactically95, although no specific studies have actually been performed to support their prophylactic use.
Hand-foot skin reaction (HFSR) can arise within the first 6 weeks of therapy, especially with kinase inhibitors targeting VEGFR, such as sunitinib and sorafenib96, but also occur with other agents targeting angiogenesis. HFSR manifests as skin blisters and callus formations that are localized on areas of pressure or friction on palms and soles. The skin changes worsen with continued therapy, and often negatively impact patients QoL due to pain and disfigurement96. Although similar in clinical presentation, HFSR is distinct from hand-foot syndrome (HFS, also known as palmar-plantar erythrodysesthesia), an undesired toxicity of various chemotherapeutic agents, such as capecitabine or 5-FU97. In HFSR, histologically, epidermal cells undergo maturation defects, while HFS is characterized by apoptotic cell changes in response to the toxic drug effects97,98. A meta-analysis of RCTs comparing the toxic effects of 5-FU based on the mode of its administration, found the incidence of HFS to be 13% when the drug was administered as bolus, and 34% when it was given as continuous infusion (P <0.0001)99. Conversely, HFSR incidence with kinase inhibitors has been reported to range between 1% and 45%. Sorafenib caused HFSR in 30% of patients treated for advanced clear-cell renal cell carcinoma, 6% of which were at grades 3 and 4 (Ref. 100), while the incidence rate with sunitinib was lower (20% of patients experienced HFSR, 5% at grades 3 and 4)101. Clinically, HFSR typically resolves within 2–4 weeks of treatment interruption, and recurs if the kinase inhibitor is reintroduced at the same dose. As no grading specifically exists for HFSR, the HFS grading is commonly used. Avoiding exposure of hands and feet to any kind of heat or friction is recommended, and to cool them with ice packs while on treatment to decrease blood flow and consequently reduce drug exposure to skin areas with a thick corneal layer and absence of sebaceous glands. Expert opinion suggests National Cancer Institute Grade 1 HFS (erythema without pain) be treated with moisturizers or emollients. Barrier repair moisturizers may reduce symptoms, as they help reduce loss of fluid and maintain/rebuild the integrity of the skin. Whether urea/lactic acid-based keratinolytics are effective, is still unclear. The rationale for recommending these agents has been their hydrating property and the ability to act as a chemical peel for hyperkeratotic skin areas. Mild itching can be managed with oatmeal baths and oral antihistamines, such as diphenhydramine. Grade 2 HFS (skin changes and/or pain) and more pronounced itching require topical corticosteroids, such as 1% hydrocortisone creams applied once or twice daily to affected areas, and topical or systemic analgesics, including opioids. Grade 3 (ulcerative dermatitis and/or pain impeding function) requires temporary interruption of kinase inhibitors and dose reduction with resumption. Other palliative therapies include pyridoxine (vitamin B6), cyclooxygenase (COX)-2 inhibitors, gabapentinoids, systemic corticosteroids, and transdermal nicotine. Despite the lack of efficacy data in the setting of kinase inhibitor therapy96,97, the same recommendations are usually followed to treat HFSR.
Inhibitors of the RAF kinase, sorafenib, vemurafenib and dabrafenib, are associated with a 6–10% incidence rate of cutaneous epithelial tumour development, such as keratoacanthomas and squamous cell carcinomas30,102. These tumours may present within a few weeks to more than 3 years from the start of treatment103,104,105. Cutaneous epithelial tumours have a low risk for metastasis and may regress spontaneously, and can be removed by surgery or cryoablation, and treatment with the respective kinase inhibitor can be continued. Moreover, several cases of basal cell carcinoma (BCC) have been reported with kinase inhibitors; however, as BCC is the most common malignant skin neoplasm, it has been difficult to correlate its development directly with kinase inhibitor use102.
TCTs can cause a variety of nonspecific skin toxicities, including xerosis, hair colour and growth changes, skin hyperpigmentation and hypopigmentation, teleangiectasias, acral erythema, and subungual haemorrhages, beginning several weeks after therapy initiation106,107. A systematic review of 18 TCT drugs found 17.4% incidence of all-grade pruritus108. Alopecia can occur with BRAF (vemurafenib, dabrafenib)109 and VEGFR inhibitors (sorafenib)110, albeit at significantly lower frequency than with conventional chemotherapy.
Wound healing and tissue repair
Kinase inhibitors and mAbs can impair wound healing and tissue repair111. This has been reported for inhibitors of the VEGF pathway (axitinib, cabozantinib, pazopanib, regorafenib, sorafenib, sunitinib, vandetanib, bevacizumab), but is documented in the labels of other agents, such as inhibitors of the ABL pathway (ponatinib). Impaired wound healing is of concern for cancer patients on treatment with TCTs, who require surgery or an invasive procedure. Due to the lack of formal studies and data on the true effects of kinase inhibitors and most mAbs on tissue repair, there are no guidelines on how to best manage these patients. Postoperative safety concerns have led to the practice of discontinuing kinase inhibitors and mAbs a few weeks before elective surgery, depending on the half-life of the agent used. This practice, however, has been questioned by some, specifically in the context of kinase inhibitor use, because the temporary suspension of a drug that has already resulted in a favourable treatment response might negatively impact the disease course112,113. Additionally, the incidence of surgery-associated complications may potentially be increased, such as the formation of intraoperative adhesions114, gastrointestinal or non-gastrointestinal perforations, fistula formations, and/or haemorrhagic or thromboembolic events. Moreover, there is no consensus on when to resume or initiate targeted agents after surgery111. In the case of bevacizumab, the recommendation is to not administer this drug for at least 28 days after a major surgery. In patients with wound dehiscence, it is recommended that treatment with bevacizumab or a kinase inhibitor is stopped.
Pulmonary toxicities
Anti-EGFR and VEGFR-directed kinase inhibitors, such as erlotinib, gefitinib and sorafenib, are reported to cause acute and potentially fatal interstitial lung disease (ILD) in approximately 1–2% of treated patients115,116. For example, ILD can develop within a median of 47 days of erlotinib treatment117. Risk factors are prior damage to the lungs, caused by radiation or chemotherapy. Symptoms are typically cough and (progressive) dyspnoea in the presence or absence of elevated body temperature. Proper pulmonary and infectious evaluation of the symptoms is mandated, as differential diagnoses include infectious causes or lymphangitic spread of the disease. Imaging of the lungs and obtaining sputum cultures are needed, and potentially bronchoscopy and broncho-alveolar lavage. In the event of ILD diagnosis, discontinuing the targeted agent and providing supportive care are critical for a patient's survival. Administration of high doses of corticosteroids and mechanical ventilation may be required. Fatality rates are high, up to 40%; the rates were particularly high with gefitinib118,119,120. Other pulmonary complications with kinase inhibitors requiring treatment with corticosteroids and discontinuation of the targeted agent may include pneumonitis121, pulmonary fibrosis122 and acute respiratory distress syndrome117.
Kinase inhibitors that target the Bcr-Abl kinase, such as dasatinib, bosutinib, imatinib and nilotinib, may cause pleural effusions123,124. The frequency is highest with dasatinib use, where 10–54% of patients are reported to be affected30,53,123. Up to 40% of patients require thoracocentesis to drain effusion volumes of more than 25% of one lung volume125. Although pleural effusions may resolve with drug cessation, as they seem to develop following daily doses higher than 100 mg or with twice daily dosing126,127, the need for hospitalization and thoracocentesis in a high percentage of patients contributes to increased financial treatment burden125.
Pulmonary toxicities related to mAbs128 range from bronchospasms that develop during drug infusion, especially common for rituximab129 (incidence rate 10%), to interstitial pneumonitis130 (incidence rate up to 7%), pulmonary fibrosis131, haemoptysis (bevacizumab- incidence rate 1.9%)132 to diffuse alveolar haemorrhage (alemtuzumab)133. Temporarily discontinuing the TCT and resuming at a lower dose or at a slower infusion rate, may be effective for acute bronchospasm and dyspnoea. In more-severe pulmonary manifestations, corticosteroid administration, termination of treatment with the targeted agent, close monitoring of the patient and aggressive management of symptoms and supportive care are essential to prevent fatality.
Renal toxicities
Renal dysfunction is common with use of anticancer drugs. This is due not only to the toxic effect of the drugs, but also because approximately 60% of patients with malignancies have an underlying kidney problem at the time of anticancer treatment134. Among the cytotoxic chemotherapeutics, renal toxicities occur primarily with cisplatin, gemcitabine, ifosphamide, methotrexate and pemetrexed. The underlying pathophysiology is diverse, and includes acute injury to the renal tubules, damage to the renal vasculature that may manifest as thrombotic microangiopathy (TMA), or crystal formation resulting in tubular damage134.
With TCTs, renal toxicities are seen especially with antiangiogenic agents, such as bevacizumab135. Toxicities range from mild to severe and can develop within 3–17 months of treatment initiation136. Manifestations include proteinuria of various degrees at a very high rate136, that can result in arterial hypertension137, TMA138, haematuria and acute renal failure. However, the symptoms generally improve with discontinuation of the TCT. Renal toxicities occur with sunitinib (creatinine elevation and hypertension)101, sorafenib (proteinuria, creatinine elevation, hypophosphataemia)139,140,141, and axitinib (hypertension, hypophosphataemia, proteinuria)142,143. Acute tubular necrosis requiring haemodialysis has been reported for imatinib144,145. Electrolyte derangements146, primarily hypomagnesaemia, occur with cetuximab and panitumumab134,147,148. It is important to determine a patient's baseline renal function and to optimize it before treatment with kinase inhibitors. Furthermore, while on treatment, patients must be monitored carefully and their renal parameters evaluated routinely and regularly. Recommended laboratory evaluations include serum creatinine and blood, urea, nitrogen (BUN) levels, serum and urine electrolytes, urine protein/albumin concentrations, and urine analysis for pH and osmolarity. Moreover, at least daily measurement of a patient's blood pressure (BP) is critical. Repletion of magnesium is best done intravenously, as oral magnesium is frequently insufficient and ineffective, and can cause diarrhoea. It is important to note that with TCTs renal toxicities can manifest at any stage of treatment. They do not seem to be dependent on the administered or the cumulative drug dose. They are, however, reversible, if precautionary and appropriate timely measures are taken149.
Neurological complications
Reversible posterior leukoencephalopathy syndrome (PRES) is seen in some cases with anti-VEGF targeting agents, especially sorafenib, sunitinib and bevacizumab52,150. This complication is rare with conventional chemotherapy, where only a few reports linked PRES to the administration of cisplatin, cyclophosphamide, L-asparaginase or gemcitabine150. PRES may be related to drug-induced hypertension or to the extent of BP change from baseline. A rise in diastolic BP is worrisome, even if only moderate, and warrantees close monitoring and prompt correction. PRES can also develop independent of hypertension, possibly due to drug effects on the brain vasculature and blood-brain barrier. Symptoms can include headaches, altered consciousness, visual changes that can range from blurred vision to cortical blindness, and even seizures150,151,152. Discontinuing the drug, performing brain imaging, ideally with MRI, and close monitoring of the patient, and managing the patient's BP and providing supportive care usually result in resolution of the symptoms153. There are no data available on whether or not the TCTs can be safely resumed after resolution of PRES.
Sunitinib, sorafenib or bevacizumab can be associated with cerebrovascular events: ischaemic or haemorrhagic strokes, intracranial haemorrhages and intracranial thromboembolic events65,66. If a patient's neurological status changes, it is essential to facilitate immediate neurological evaluation. Sensory neuropathy can be frequent and cumulative with platinum-based chemotherapy, taxanes and vinca alkaloids, but develops only very rarely with the use of some mAbs, such as bevacizumab, brentuzimab-vidotin and trastuzumab152. With trastuzumab, symptoms can develop with a delay of 1 year after treatment initiation, likely indicating a cumulative effect of the drug152. If the neuropathy is painful, pain relievers (NSAIDs, opioids), anti-seizure medications (gabapentin, pregabalin) and various anti-depressants can be prescribed.
Muscle cramping, myalgias and sometimes serum creatine phosphokinase (CPK) elevations are reported for imatinib151. They occur in 20–49% of patients, are usually mild, and respond well to calcium and magnesium treatment, or to quinine151. Cognitive dysfunction without direct CNS involvement by the underlying malignancy is not commonly seen with kinase inhibitors or mAbs52. There are reports on sunitinib-associated cognitive disorder (confusion, hallucinations, and extrapyramidal syndrome) with doses of 50 mg in patients older than 70 years and with pre-existing arteriosclerotic leukoencephalopathy154. These symptoms resolved rapidly with discontinuation of the drug.
Endocrine side effects
Hypothyroidism or hyperthyroidism can occur within weeks of kinase inhibitor treatment30,155,156. The prevalence of hypothyroidism in patients treated with these agents varies depending on the drug used and ranges from 3% to 89%155,156. Hypothyroidism is common with sunitinib; retrospective studies indicate that 53–83% of patients receiving the drug can become hypothyroid, while a lower range has been observed (36–71%) in prospective studies157,158,159. Sunitinib, sorafenib and axitinib are associated with hyperthyroidism and thyrotoxicosis, although the reported prevalence is lower than for hypothyroidism160,161,162,163. Sunitinib and imatinib can increase the requirement for thyroid hormone supplementation in patients with pre-existing hypothyroidism164,165. Several potential mechanisms have been proposed, and include increased activity of type 3 iodothyronine deiodinase (D3)166,167, the physiological inactivator of thyroid hormones168, regression of thyroidal capillaries due to inhibition of angiogenesis169, impaired iodine uptake170, and hypothyroidism as a consequence of thyroiditis with transient thyrotoxicosis160,162. Another possible reason for hypothyroidism with bosutinib and dasatinib is the inhibition of the thyroid hormone transporter MCT8 (Ref. 171). The resulting lack of thyroid hormone feedback to the hypothalamus/pituitary axis can elevate blood levels of thyroid stimulating hormone. A major concern with kinase inhibitor-induced thyroid dysfunction is its impact on the QoL of patients, as it may cause or aggravate fatigue155. Supplementation with thyroid hormones to normalize thyroid stimulating hormone levels can help alleviate symptoms155, but concern has been raised about potential tumour growth with thyroid hormone replacement in patients with active cancers172.
Ocular toxicities
Ocular toxicities with targeted agents range from mild symptoms, such as blurred vision, altered light perception, conjunctivitis, to severe vision impairement, such as corneal erosion, ulcer, perforation, optic neuritis, vitreous haemorrhage, retinal detachment, and retinal vascular occlusion173. Most EGFR inhibitors cause blepharitis and lacrimation disorders, with the exception of lapatinib, for which no ocular adverse effects have been reported25,165,166. C-kit inhibitors, which include imatinib, pazopanib, sunitinib, can cause orbital and periorbital oedema, although systemic use of VEGF pathway inhibitors rarely cause direct ocular defects30,173,174. The visual syndromes observed with VEGF/VEGFR inhibitors tend to be related to cardiovascular or thromboembolic toxicities174.
Overall, the reported incidence rates and degrees of severity of ocular adverse effects vary substantially in the reports of clinical trials compared to those reported in drug labels. Nonetheless, every structure of the eye, including the eyelids, can be affected, and patients might experience pain. As patients with pre-existing ophthalmologic conditions are at higher risk of developing ocular adverse effects, particular caution and close monitoring is advised. Prompt recognition of symptoms and ophthalmologic evaluation are also indicated174.
Additional challenges
As kinase inhibitors and mAbs continue to play an essential role in contemporary cancer therapy, it is difficult to interpret the results reported in oncology trials and ascertain the benefits and risks of these drugs. With data indicating expanding utilization of anticancer drugs worldwide175, a better understanding of the effects of TCTs is required to maximize their therapeutic potential and reduce negative impacts on patient daily functioning and QoL176. Given that most kinase inhibitors are administered orally and in the outpatient setting, often for long time periods, information about treatment-related adverse events including possible cumulative toxicities and their impact on QoL may be best achieved through patient-reported data177,178.
Cost-effectiveness and symptom burden
In the USA, debate persists around the cost-effectiveness of many kinase inhibitors and mAbs in relation to agreed-upon standards of efficacy, such as PFS and overall survival179. Direct costs associated with TCTs are significant180; for instance, 1 year of treatment with trastuzumab can exceed US$50,000, and with pertuzumab more than $70,000. This cost is low compared to the annual costs of the latest immunotherapy drugs that are approximately $120,000 annual cost for ipilumumab, $150,000 for pembrolizumab, or $178,000 for blinatumomab. Although all or part of the costs may be covered by insurance, the expenses still represent a burden to the overall health care costs, apart from the fact that frequently patients do not have any insurance coverage. A growing older population coupled with newer, more sophisticated therapies will undoubtedly drive the overall cost of cancer therapy in coming decades181. Associated indirect costs, such as mutation screening, contribute to discussions about therapy costs and appropriate resource utilization182. Although generally considered to be better tolerated than conventional chemotherapies, the clinical benefit offered by TCTs is frequently only moderate179, and the median overall survival gain over conventional chemotherapy has been described to be 2 months183. The economic cost associated with toxicities and symptom burden in relation to true clinical benefit from TCTs is just beginning to be evaluated184,185. Future research should continue to determine incremental cost-effectiveness of specific TCTs in relation to their therapeutic potential and clinical benefit186.
End-of-life care
The increased use of TCTs in standard cancer management and their oral administration show that more patients are choosing to continue these pharmacotherapies late in the disease course; this situation poses new challenges for appropriate care at the end of life187. In the past, conventional chemotherapy use in the final weeks of life has been associated with decreased likelihood of hospice use in the USA188. Many patients might forego the harsher toxicities of conventional chemotherapy and be persuaded toward comfort-focused care. Indeed, administration of chemotherapy within the last 2–4 weeks of life, and late referral to hospice services, remain negative quality-of-care indicators189. However, the lighter symptom burden generally purported by TCTs and their oral administration mode may facilitate additional attempts at disease-modifying therapies, despite the low likelihood of benefit190,191.
This phenomenon may delay or even prevent hospice enrolment for several reasons. First, the increased treatment options for patients (and oncologists) who wish to pursue additional attempts at disease modifying therapy, however little the likelihood of success. Second, patients who would otherwise be ineligible for conventional chemotherapies can continue oral formulations of kinase inhibitors at home192. In the USA, current hospice enrolment conditions of participation and reimbursement mechanisms can create barriers for patients who may need hospice services, but wish to continue disease-modifying therapies. The Medicare Hospice Benefit (MHB), which provides financial stability to the vast majority of hospice care in the USA, requires that patients exchange their regular Medicare coverage for the terminal condition for hospice services, effectively disallowing ongoing life-sustaining therapies193. The MHB reimburses hospice agencies on a per-diem basis to cover all expenses related to the hospice diagnosis (including pharmacotherapies); this amount (approximately $150 per day for routine home care) coupled with an annual fiscal cap on total payments to each hospice attempt to control hospice expenditures193. Given reimbursement limitations, most US hospices elect enrolment policies to restrict access to care for patients with potentially high costs, including chemotherapeutics or TCTs194. Faced with this choice between supportive hospice care and continuing potentially-life sustaining therapies, many cancer patients who choose to forego hospice, may in fact still benefit from the hospice support that they declined195. The decision of some patients and their caregivers to continue treatment with targeted agents is often rooted in unrealistic expectations that these drugs may miraculously help 'cure' or 'halt' growth of the malignancy. This is not surprising considering distorted media reports of 'breakthroughs' in cancer research and treatment, aggressive industry promotions of drugs as well as biased reporting of clinical trial results175. In addition, the increasing patient diagnoses, and relatively short time that physicians have to devote to each patient, treatment goals and patients' preferences for end-of-life care may not be addressed adequately and/or in a timely manner, so that the value of palliative and hospice care at an advanced stage of the disease and at the final stage of life may not always be evident to patients and their caregivers196. Negative implications of this trend include less time for hospice teams to establish crucial relationships with patients and caregivers, and the potential for greater symptom burden due to toxicities from TCT. Moreover, terminal hospitalizations of cancer patients versus hospice utilization do not only impact the patient's physical and emotional wellbeing, but also the mental health status of the patients' caregiver(s)197. Patients who died in the ICU or the hospital setting was found to be associated with increased rates of bereavement-related psychiatric illnesses in caregivers compared to those, whose family member died at home with hospice care197. A recent study reported that almost one third of patients with cancer have hospice enrolment periods of less than 1 week198, underlining the above-mentioned barriers and confirming previous reports199. In addition, statistically significant differences exist in care utilization between non-hospice and hospice Medicare fee-for-service beneficiaries with a poor prognosis200. Lower rates of hospitalizations, ICU admissions and invasive procedures significantly reduce health care expenditures during the last year of life for those enrolled in hospice care compared to matched controls not receiving hospice care200. The greatest savings are produced with hospice enrolment of 5–8 weeks supporting the benefit of earlier hospice enrolment for both the individual patient and the healthcare system.
Conclusions
The advent of kinase inhibitors and mAbs provide a meaningful revolution in managing malignant diseases. Although the use of some of these drugs has resulted in a clinical benefit for patients, especially in earlier disease settings, some even outperforming conventional chemotherapy, such as erlotinib201 and crizotinib202, the successes have been associated with important unforeseen implications. Designed to specifically target the malignant cell, normal cells with similar molecular targets remain poised to be affected by collateral damage. Despite hopes of minimal drug toxicities, these novel agents are associated with undesired effects that demand physicians' awareness to place the risks into the correct context of potential clinical benefit. The significance of this is underlined by the fact that kinase inhibitors are given orally and for prolonged time periods, frequently during advanced-stage disease and the final stage of life. Hence, it is imperative to understand and appropriately address any undesired drug effects, as treatment-related toxicities can prove to be debilitating and costly to the patient and the healthcare system. Particular challenges in this context are the correct recognition of adverse events that reflect good response to treatment (such as skin rash with EGFR inhibitors or hypertension with angiogenesis inhibitors), as opposed to those that do not correlate with treatment efficacy, because the management approaches can vary. In addition, it is unclear when to reasonably stop treatment in advanced-stage diseases and at the end of life to avoid harming patients and negatively impacting their QoL175,179,196. With increasing clinical use of these pharmacotherapies, greater patient-centred research into management of toxicities and QoL outcomes is warranted. Collectively, such efforts should prove beneficial in providing better care throughout the disease trajectory for the growing populations living with cancer. In addition, with the development of new agents, such as bispecific T-cell engaging antibodies, chimeric antigen receptor T-cell therapies, one can anticipate a rise in new and unique toxicities, including cytokine-release syndrome and neurological toxicities. It will be important for oncologists to stay updated on the latest developments and drug approvals, as the field advances rapidly.
References
Krause, D. S. & Van Etten, R. A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 353, 172–187 (2005).
Amir, E. et al. Oncogenic targets, magnitude of benefit, and market pricing of antineoplastic drugs. J. Clin. Oncol. 29, 2543–2549 (2011).
DiMasi, J. A. & Grabowski, H. G. Economics of new oncology drug development. J. Clin. Oncol. 25, 209–216 (2007).
Piccart-Gebhart, M. J. et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 353, 1659–1672 (2005).
Slamon, D. J. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792 (2001).
Druker, B. J. et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 355, 2408–2417 (2006).
Baldo, B. A. & Pham, N. H. Adverse reactions to targeted and non-targeted chemotherapeutic drugs with emphasis on hypersensitivity responses and the invasive metastatic switch. Cancer Metastasis Rev. 32, 723–761 (2013).
Niraula, S. et al. The price we pay for progress: a meta-analysis of harms of newly approved anticancer drugs. J. Clin. Oncol. 30, 3012–3019 (2012).
Funakoshi, T., Latif, A. & Galsky, M. D. Safety and efficacy of addition of VEGFR and EGFR-family oral small-molecule tyrosine kinase inhibitors to cytotoxic chemotherapy in solid cancers: a systematic review and meta-analysis of randomized controlled trials. Cancer Treat. Rev. 40, 636–647 (2014).
Yancik, R., Ganz, P. A., Varricchio, C. G. & Conley, B. Perspectives on comorbidity and cancer in older patients: approaches to expand the knowledge base. J. Clin. Oncol. 19, 1147–1151 (2001).
Pajares, B. et al. Tyrosine kinase inhibitors and drug interactions: a review with practical recommendations. Clin. Transl. Oncol. 14, 94–101 (2012).
Hartmann, J. T., Haap, M., Kopp, H. G. & Lipp, H. P. Tyrosine kinase inhibitors — a review on pharmacology, metabolism and SEs. Curr. Drug Metab. 10, 470–481 (2009).
Imai, K. & Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 6, 714–727 (2006).
Dassonville, O., Bozec, A., Fischel, J. L. & Milano, G. EGFR targeting therapies: monoclonal antibodies versus tyrosine kinase inhibitors: similarities and differences. Crit. Rev. Oncol. Hematol. 62, 53–61 (2007).
Chari, R. V. J. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc. Chem. Res. 41, 98–107 (2008).
Köhler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975).
Carter, P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 1, 118–129 (2001).
Beck, A., Wurch, T. & Corvaia, N. Therapeutic antibodies and derivatives: from the bench to the clinic. Curr. Pharm. Biotechnol. 9, 421–422 (2008).
Nelson, A. L., Dhimolea, E. & Reichert, J. M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 9, 767–774 (2010).
Oldham, R. K. & Dillman, R. O. Monoclonal antibodies in cancer therapy: 25 years of progress. J. Clin. Oncol. 26, 1774–1777 (2008).
Nissim, A. & Chernajovsky, Y. Historical development of monoclonal antibody therapeutics. Handb. Exp. Pharmacol. 181, 3–18 (2008).
Yamada, T. Therapeutic monoclonal antibodies. Keio J. Med. 60, 37–46 (2011).
Scott, A. M., Wolchok, J. D. & Old, L. J. Antibody therapy of cancer. Nat. Rev. Cancer 12, 278–287 (2012).
Widmer, N. et al. Review of therapeutic drug monitoring of anticancer drugs part two — targeted therapies. Eur. J. Cancer 50, 2020–2036 (2014).
Huang, S. et al. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 64, 5355–5362 (2004).
Lee, S. J. & Kavanaugh, A. Adverse reactions to biologic agents: focus on autoimmune disease therapies. J. Allergy Clin. Immunol. 116, 900–905 (2005).
Pichler, W. J. Adverse SEs to biologic agent. Allergy 61, 912–920 (2006).
Albini, A. et al. Cardiotoxicity of anticancer drugs: the need for cardio-oncology and cardio-oncological prevention. J. Natl Cancer Inst. 102, 14–25 (2010).
O'Brien, S. G. et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 348, 994–1004 (2003).
Dy, G. K. & Adjei, A. A. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA Cancer J. Clin. 63, 249–279 (2013).
Lambertini, M., Peccatori, F. A. & Azim, H. A. Jr. Targeted agents for cancer treatment during pregnancy. Cancer Treat. Rev. 41, 301–309 (2015).
Riechelmann, R. P. et al. Potential drug interactions and duplicate prescriptions among cancer patients. J. Natl Cancer Inst. 99, 592–600 (2007).
Reya, T., Morrison, S. J., Clarke, M. F. & Weissman, I. L. Stem cells, cancer, and cancer stem cells. Nature 414, 105–111 (2001).
Dean, M., Fojo, T. & Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 5, 275–284 (2005).
Reslan, L., Dalle, S. & Dumontet, C. Understanding and circumventing resistance to anticancer monoclonal antibodies. MAbs 1, 222–229 (2009).
Perez-Soler, R. Rash as a surrogate marker for efficacy of epidermal growth factor receptor inhibitors in lung cancer. Clin. Lung Cancer 8, S7–S14 (2006).
Chen, M. H. Cardiac dysfunction induced by novel targeted anticancer therapy: an emerging issue. Curr. Cardiol. Rep. 11, 167–174 (2009).
Ewer, M. S. et al. Reversibility of trastuzumab-related cardiotoxicity: new insights based on clinical course and response to medical treatment. J. Clin. Oncol. 23, 7820–7826 (2005).
Force, T., Krause, D. S. & Van Etten, R. A. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 7, 332–344 (2007).
Chen, M. H., Kerkelä, R. & Force, T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 118, 84–95 (2008).
Shakir, D. K. & Rasul, K. I. Chemotherapy induced cardiomyopathy: pathogenesis, monitoring and management. J. Clin. Med. Res. 1, 8–12 (2009).
Seidman, A. et al. Cardiac dysfunction in the trastuzumab clinical trials experience. J. Clin. Oncol. 20, 1215–1221 (2002).
Bria, E. et al. Cardiotoxicity and incidence of brain metastases after adjuvant trastuzumab for early breast cancer: the dark side of the moon? A meta-analysis of the randomized trials. Breast Cancer Res. Treat. 109, 231–239 (2008).
Perez, E. A. et al. Cardiac safety of lapatinib: pooled analysis of 3689 patients enrolled in clinical trials. Mayo Clin. Proc. 83, 679–686 (2008).
Chen, T. et al. Risk of cardiac dysfunction with trastuzumab in breast cancer patients: a meta-analysis. Cancer Treat. Rev. 37, 312–320 (2011).
Lenihan, D. et al. Pooled analysis of cardiac safety in patients with cancer treated with pertuzumab. Ann. Oncol. 23, 791–800 (2012).
Svoboda, M. et al. Cardiac toxicity of targeted therapies used in the treatment for solid tumours: a review. Cardiovasc. Toxicol. 12, 191–207 (2012).
Chu, T. F. et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 370, 2011–2019 (2007).
Schmidinger, M. et al. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 26, 5204–5212 (2008).
Shah, R. R., Morganroth, J. & Shah, D. R. Cardiovascular safety of tyrosine kinase inhibitors: with a special focus on cardiac repolarisation (QT interval). Drug Saf. 36, 295–316 (2013).
Vaughn, C., Zhang, L. & Schiff, D. Reversible posterior leukoencephalopathy syndrome in cancer. Curr. Oncol. Rep. 10, 86–91 (2008).
Lee, E. Q., Arrillaga-Romany, I. C. & Wen, P. Y. Neurologic complications of cancer drug therapies. Continuum (Minneap. Minn.) 18, 355–365 (2012).
Quintás-Cardama, A. et al. Pleural effusion in patients with chronic myelogenous leukemia treated with dasatinib after imatinib failure. J. Clin. Oncol. 25, 3908–3914 (2007).
Montani, D. et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 125, 2128–2137 (2012).
Rothenstein, J. M. & Letarte, N. Managing treatment-related adverse events associated with Alk inhibitors. Curr. Oncol. 21, 19–26 (2014).
Ou, S. H., Azada, M., Dy, J. & Stiber, J. A. Asymptomatic profound sinus bradycardia (heart rate ≤45) in non-small cell lung cancer patients treated with crizotinib. J. Thorac. Oncol. 6, 2135–2137 (2011).
Ou, S. H. et al. Heart rate decrease during crizotinib treatment and potential correlation to clinical response. Cancer 119, 1969–1975 (2013).
Scappaticci, F. A. et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J. Natl Cancer Inst. 99, 1232–1239 (2007); erratums 100, 156 (2008), 100, 685 (2008).
Grivas, A. A., Trafalis, D. T. & Athanassiou, A. E. Implication of bevacizumab in fatal arterial thromboembolic incidents. J. BUON 14, 115–117 (2009).
Patel, J. N. et al. Bevacizumab and the risk of arterial and venous thromboembolism in patients with metastatic, castration-resistant prostate cancer treated on Cancer and Leukemia Group B (CALGB) 90401 (Alliance). Cancer 121, 1025–1031 (2015).
Hurwitz, H. I. et al. Venous thromboembolic events with chemotherapy plus bevacizumab: a pooled analysis of patients in randomized phase II and III studies. J. Clin. Oncol. 29, 1757–1764 (2011).
Nalluri, S. R. et al. Risk of venous thromboembolism with the angiogenesis inhibitor bevacizumab in cancer patients: a meta-analysis. JAMA 300, 2277–2285 (2008).
Chu, D. & Wu, S. Risk of venous thromboembolism with bevacizumab in cancer patients — reply. JAMA 301, 1435–1436 (2009).
Ferroni, P., Formica, V., Roselli, M. & Guadagni, F. Thromboembolic events in patients treated with anti-angiogenic drugs. Curr. Vasc. Pharmacol. 8, 102–113 (2010).
Schutz, F. A. et al. Bevacizumab increases the risk of arterial ischemia: a large study in cancer patients with a focus on different subgroup outcomes. Ann. Oncol. 22, 1404–1412 (2011).
Choueiri, T. K. et al. Risk of arterial thromboembolic events with sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. J. Clin. Oncol. 28, 2280–2285 (2010).
Sonpavde, G. et al. Venous thromboembolic events with vascular endothelial growth factor receptor tyrosine kinase inhibitors: a systematic review and meta-analysis of randomized clinical trials. Crit. Rev. Oncol. Hematol. 87, 80–89 (2013).
Kantarjian, H. et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 362, 2260–2270 (2010).
Clark, O. A. et al. Colony-stimulating factors for chemotherapy-induced febrile neutropenia: a meta-analysis of randomized controlled trials. J. Clin. Oncol. 23, 4198–4214 (2005).
Barber, N. A., Afzal, W. & Akhtari, M. Hematologic toxicities of small molecule tyrosine kinase inhibitors. Target Oncol. 6, 203–215 (2011).
Gibson, R. J. & Stringer, A. M. Chemotherapy-induced diarrhoea. Curr. Opin. Support. Palliat. Care 3, 31–35 (2009).
Stein, A., Voigt, W. & Jordan, K. Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management. Ther. Adv. Med. Oncol. 2, 51–63 (2010).
Lu, Y. Y. et al. Clinical observations on associations between the UGT1A1 genotype and severe toxicity of irinotecan. Asian Pac. J. Cancer Prev. 15, 3335–3341 (2014).
Loriot, Y. et al. Drug insight: gastrointestinal and hepatic adverse effects of molecular-targeted agents in cancer therapy. Nat. Clin. Pract. Oncol. 5, 268–278 (2008).
Thomas, S. K. et al. Asian ethnicity as a predictor of response in patients with non-small-cell lung cancer treated with gefitinib on an expanded access program. Clin. Lung Cancer 7, 326–331 (2006).
Kanazawa, S. et al. Aspirin reduces adverse effects of gefitinib. Anticancer Drugs 17, 423–427 (2006).
Demetri, G. D. et al. Efficacity and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 347, 472–480 (2002).
Van Glabbeke, M. et al. Predicting toxicities for patients with advanced gastrointestinal stromal tumours treated with imatinib: a study of the European Organisation for Research and Treatment of Cancer, the Italian Sarcoma Group, and the Australasian Gastro-Intestinal Trials Group (EORTC ISG-AGITG). Eur. J. Cancer 42, 2277–2278 (2006).
Uribe, J. M., Gelbmann, C. M., Traynor-Kaplan, A. E. & Barrett, K. E. Epidermal growth factor inhibits Ca2+-dependent Cl− transport in T84 human colonic epithelial cells. Am. J. Physiol. 271, C914–922 (1996).
Melosky, B. Supportive care treatments for toxicities of anti-EGFR and other targeted agents. Curr. Oncol. 19, S59–S63 (2012).
Hirsh, V. et al. Management of diarrhea induced by epidermal growth factor receptor tyrosine kinase inhibitors. Curr. Oncol. 21, 329–336 (2014).
Ohyashiki, K. et al. Imatinib mesylate-induced hepato-toxicity in chronic myeloid leukemia demonstrated focal necrosis resembling acute viral hepatitis. Leukemia 16, 2160–2161 (2002).
James, C. et al. Histological features of acute hepatitis after imatinib mesylate treatment. Leukemia 14, 978–979 (2003).
Kikuchi, S. et al. Severe hepatitis and complete molecular response caused by imatinib mesylate: possible association of its serum concentration with clinical outcomes. Leuk. Lymphoma 45, 2349–2351 (2004).
Cross, T. J. S. et al. Imatinib mesylate as a cause of acute liver failure. Am. J. Hematol. 81, 189–192 (2006).
Pariente, A. et al. Imatinib mesylate-induced acute hepatitis in a patient treated for gastrointestinal stromal tumour. Eur. J. Gastroenterol. Hepatol. 18, 785–787 (2006).
Dhalluin-Venier, V. et al. Imatinib mesylate induced acute hepatitis with autoimmune features. Eur. J. Gastroenterol. Hepatol. 18, 1235–1237 (2006).
Srinivasan, S., Parsa, V., Liu, C. Y. & Fontana, J. A. Trastuzumab-induced hepatotoxicity. Ann. Pharmacother. 42, 1497–1501 (2008).
Pessaux, P. et al. Targeted molecular therapies (cetuximab and bevacizumab) do not induce additional hepatotoxicity: preliminary results of a case-control study. Eur. J. Surg. Oncol. 36, 575–582 (2010).
Lin, N. U. et al. Fatal hepatic necrosis following imatinib mesylate therapy. Blood 102, 3455–3456 (2003).
Robert, C. et al. Cutaneous side-effects of kinase inhibitors and blocking antibodies. Lancet Oncol. 6, 491–500 (2005).
Joshi, S. S. et al. Effects of epidermal growth factor receptor inhibitor-induced dermatologic toxicities on quality of life. Cancer 116, 3916–3923 (2010).
Jatoi, A. et al. Clinical predictors of severe cetuximab-induced rash: observations from 933 patients enrolled in North Central Cancer Treatment Group Study N0147. Oncology 77, 120–123 (2009).
Baas, J. M. et al. Recommendations on management of EGFR inhibitor-induced skin toxicity: a systematic review. Cancer Treat. Rev. 38, 505–514 (2012).
Bachet, J. B. et al. Folliculitis induced by EGFR inhibitors, preventive and curative efficacy of tetracyclines in the management and incidence rates according to the type of EGFR inhibitor administered: a systematic literature review. Oncologist 17, 555–568 (2012).
Lipworth, A. D., Robert, C. & Zhu, A. X. Hand-foot syndrome (hand-foot skin reaction, palmar-plantar erythrodysesthesia): focus on sorafenib and sunitinib. Oncology 77, 257–271 (2009).
Nagore, E., Insa, A. & Sanmartín, O. Antineoplastic therapy-induced palmar plantar erythrodysesthesia ('hand-foot') syndrome: ncidence, recognition and management. Am. J. Clin. Dermatol. 1, 225–234 (2000).
Segaert, S. et al. Skin toxicities of targeted therapies. Eur. J. Cancer 45 (Suppl. 1), 295–308 (2009).
Lévy, E. et al. Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. Meta-Analysis Group In Cancer. J. Clin. Oncol. 16, 3537–3541 (1998).
Escudier, B. et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 356, 125–134 (2007); erratum 357, 203 (2007).
Motzer, R. J. et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 356, 115–124 (2007).
Breaker, K. et al. Skin cancer associated with the use of sorafenib and sunitinib for renal cell carcinoma. Dermatol. Surg. 39, 981–987 (2013).
Arnault, J. P. et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J. Clin. Oncol. 27, e59–e61 (2009).
Chapman, P. B. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 (2011).
Hauschild, A. et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380, 358–365 (2012).
Lacouture, M. E., Boerner, S. A. & Lorusso, P. M. Non-rash skin toxicities associated with novel targeted therapies. Clin. Lung Cancer 8, S36–S42 (2006).
Kyllo, R. L. & Anadkat, M. J. Dermatologic adverse events to chemotherapeutic agents, part 1: cytotoxics, epidermal growth factor receptors, multikinase inhibitors, and proteasome inhibitors. Semin. Cutan. Med. Surg. 33, 28–39 (2014).
Ennslin, C. J., Rosen, A. C., Wu, S. & Lacouture, M. Pruritus in patients treated with targeted cancer therapies: systematic review and meta-analysis. J. Am. Acad. Dermatol. 69, 708–720 (2013).
Vanneste, L. et al. Cutaneous adverse effects of BRAF inhibitors in metastatic malignant melanoma, a prospective study in 20 patients. J. Eur. Acad. Dermatol. Venereol. 29, 61–68 (2015).
Yano, K., Brown, L. F. & Detmar, M. Control of hair growth and follicle size by VEGF-mediated angiogenesis. J. Clin. Invest. 107, 409–417 (2001).
Shah, D. R., Dholakia, S. & Shah, R. R. Effect of tyrosine kinase inhibitors on wound healing and tissue repair: implications for surgery in cancer patients. Drug Saf. 37, 135–149 (2014).
Johannsen, M. et al. Can tyrosine kinase inhibitors be discontinued in patients with metastatic renal cell carcinoma and a complete response to treatment? A multicentre, retrospective analysis. Eur. Urol. 55, 1430–1439 (2009).
Johannsen, M. et al. Outcome of treatment discontinuation in patients with metastatic renal cell carcinoma and no evidence of disease following targeted therapy with or without metastasectomy. Ann. Oncol. 22, 657–663 (2011).
Harshman, L. C. et al. Surgical outcomes and complications associated with presurgical tyrosine kinase inhibition for advanced renal cell carcinoma (RCC). Urol. Oncol. 31, 379–385 (2013).
Qi, W. X., Sun, Y. J., Shen, Z. & Yao, Y. Risk of interstitial lung disease associated with EGFR-TKIs in advanced non-small-cell lung cancer: a meta-analysis of 24 phase III clinical trials. J. Chemother. 27, 40–51 (2015).
Myung, H. J. et al. Sorafenib-induced interstitial pneumonitis in a patient with hepatocellular carcinoma: a case report. Gut Liver 4, 543–546 (2010).
Barber, N. A. & Ganti, A. K. Pulmonary toxicities from targeted therapies: a review. Target Oncol. 6, 235–243 (2011).
Inoue, A. et al. Severe acute interstitial pneumonia and gefitinib. Lancet 361, 137–139 (2003).
Danson, S., Blackhall, F., Hulse, P. & Ranson, M. Interstitial lung disease in lung cancer: separating disease progression from treatment effects. Drug Saf. 28, 103–113 (2005).
Suzuki, M. et al. Recurrent gefitinib-induced interstitial lung disease. Intern. Med. 47, 533–536 (2008).
Seidel, C. et al. Recall pneumonitis during systemic treatment with sunitinib. Ann. Oncol. 21, 2119–2120 (2010).
Grimminger, F., Günther, A. & Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 45, 1426–1433 (2015).
Kantarjian, H. et al. Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia after failure of first-line imatinib: a randomized phase 2 trial. Blood 109, 5143–5150 (2007).
Khoury, H. J. et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood 119, 3403–3412 (2012).
Stephens, J., Carpiuc, K. T. & Botteman, M. The burden of managing pleural effusions in patients with chronic myelogenous leukemia post-imatinib failure: a literature-based economic analysis. Int. J. Gen. Med. 3, 31–36 (2010).
Shah, N. P. et al. Intermittent target inhibition with dasatinib 100 mg once daily preserves efficacy and improves tolerability in imatinib-resistant and -intolerant chronic-phase chronic myeloid leukemia. J. Clin. Oncol. 26, 3204–3212 (2008).
Kim, D. et al. Long-term pattern of pleural effusion from chronic myeloid leukemia patients in second-line dasatinib therapy. Int. J. Hematol. 94, 361–371 (2011).
Peerzada, M. M., Spiro, T. P. & Daw, H. A. Pulmonary toxicities of biologics: a review. Anticancer Drugs 21, 131–139 (2010).
Bouldouyre, M. A., Cohen, P. & Guillevin, L. Severe bronchospasm associated with rituximab for refractory Churg–Strauss syndrome. Ann. Rheum. Dis. 68, 606 (2009).
Burton, C., Kaczmarski, R. & Jan-Mohamed, R. Interstitial pneumonitis related to rituximab therapy. N. Eng. J. Med. 348, 2690–2691 (2003).
Leon, R. J., Gonsalvo, A., Salas, R. & Hidalgo, N. C. Rituximab-induced acute pulmonary fibrosis. Mayo Clin. Proc. 79, 953 (2004).
Johnson, D. H. et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 22, 2184–2191 (2004).
Sachdeva, A. & Matuschak, G. M. Diffuse alveolar hemorrhage following alemtuzumab. Chest 133, 1476–1478 (2008).
Perazella, M. A. Onco-nephrology: renal toxicities of chemotherapeutic agents. Clin. J. Am. Soc. Nephrol. 7, 1713–1721 (2012).
Gordon, M. S. et al. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J. Clin. Oncol. 19, 843–850 (2001).
Eremina, V. et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 358, 1129–1136 (2008).
Zhu, X., Wu, S., Dahut, W. L. & Parikh, C. R. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and meta-analysis. Am. J. Kidney Dis. 49, 186–193 (2007).
Bollée, G. et al. Thrombotic microangiopathy secondary to VEGF pathway inhibition by sunitinib. Nephrol. Dial. Transplant. 24, 682–685 (2009).
Abou-Alfa, G. K. et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 24, 4293–4300 (2006).
Chi, K. N. et al. A phase II study of sorafenib in patients with chemo-naive castration-resistant prostate cancer. Ann. Oncol. 19, 746–751 (2008).
Llovet, J. M. et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008).
Launay-Vacher, V. & Deray, G. Hypertension and proteinuria: a class-effect of antiangiogenic therapies. Anticancer Drugs 20, 81–82 (2009).
Izzedine, H. et al. VEGF signalling inhibition-induced proteinuria: mechanisms, significance and management. Eur. J. Cancer 46, 439–448 (2010).
Pou, M. et al. Acute renal failure secondary to imatinib mesylate treatment in chronic myeloid leukemia. Leuk. Lymphoma 44, 1239–1241 (2003).
Foringer, J. R. et al. Acute renal failure secondary to imatinib mesylate treatment in prostate cancer. Ann. Pharmacother. 39, 2136–2138 (2005).
Dahut, W. L. et al. A phase II clinical trial of sorafenib in androgen-independent prostate cancer. Clin. Cancer Res. 14, 209–214 (2008).
Schrag, D., Chung, K. Y., Flombaum, C. & Saltz, L. Cetuximab therapy and symptomatic hypomagnesemia. J. Natl Cancer Inst. 97, 1221–1224 (2005).
Cao, Y. et al. Meta-analysis of incidence and risk of hypomagnesemia with cetuximab for advanced cancer. Chemotherapy 56, 459–465 (2010).
Kelly, R. J., Billemont, B. & Rixe, O. Renal toxicity of targeted therapies. Target Oncol. 4, 121–133 (2009).
Marinella, M. A. & Markert, R. J. Reversible posterior leucoencephalopathy syndrome associated with anticancer drugs. Intern. Med. J. 39, 826–834 (2009).
Schiff, D., Wen, P. Y. & van den Bent, M. J. Neurological adverse effects caused by cytotoxic and targeted therapies. Nat. Rev. Clin. Oncol. 6, 596–603 (2009).
Soffietti, R., Trevisan, E. & Rudà, R. Neurologic complications of chemotherapy and other newer and experimental approaches. Handb. Clin. Neurol. 121, 1199–1218 (2014).
Bartynski, W. S. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am. J. Neuroradiol 29, 1036–1042 (2008).
van der Veldt, A. A. et al. Reversible cognitive disorders after sunitinib for advanced renal cell cancer in patients with preexisting arteriosclerotic leukoencephalopathy. Ann. Oncol. 18, 1747–1750 (2007).
Torino, F. et al. Hypothyroidism related to tyrosine kinase inhibitors: an emerging toxic effect of targeted therapy. Nat. Rev. Clin. Oncol. 6, 219–228 (2009).
Fallahi, P. et al. Thyroid dysfunctions induced by tyrosine kinase inhibitors. Expert Opin. Drug Saf. 13, 723–733 (2014).
Wolter, P. et al. The clinical implications of sunitinib-induced hypothyroidism: a prospective evaluation. Br. J. Cancer 99, 448–454 (2008).
Shinohara, N. et al. The incidence and mechanism of sunitinib-induced thyroid atrophy in patients with metastatic renal cell carcinoma. Br. J. Cancer 104, 241–247 (2011).
Funakoshi, T. & Shimada, Y. J. Risk of hypothyroidism in patients with cancer treated with sunitinib: a systematic review and meta-analysis. Acta Oncol. 52, 691–702 (2013).
Grossmann, M., Premaratne, E., Desai, J. & Davis, I. D. Thyrotoxicosis during sunitinib treatment for renal cell carcinoma. Clin. Endocrinol. (Oxf.) 69, 669–672 (2008).
Tamaskar, I. et al. Thyroid function test abnormalities in patients with metastatic renal cell carcinoma treated with sorafenib. Ann. Oncol. 19, 265–268 (2008).
Sato, S. et al. Clinical characteristics of thyroid abnormalities induced by sunitinib treatment in Japanese patients with renal cell carcinoma. Endocr. J. 57, 873–880 (2010).
Konca Degertekin, C. et al. Hyperthyroidism and thyroid autoimmunity induced by sorafenib in metastatic renal cell cancer. Endocrine 42, 756–757 (2012).
de Groot, J. W. et al. Imatinib induces hypothyroidism in patients receiving levothyroxine. Clin. Pharmacol. Ther. 78, 433–438 (2005).
Rini, B. I. et al. Hypothyroidism in patients with metastatic renal cell carcinoma treated with sunitinib. J. Natl Cancer Inst. 99, 81–83 (2007).
Abdulrahman, R. M. et al. Sorafenib-induced hypothyroidism is associated with increased type 3 deiodination. J. Clin. Endocrinol. Metab. 95, 3758–3762 (2010).
Kappers, M. H. et al. Sunitinib-induced hypothyroidism is due to induction of type 3 deiodinase activity and thyroidal capillary regression. J. Clin. Endocrinol. Metab. 96, 3087–3094 (2011).
Huang, S. A. Physiology and pathophysiology of type 3 deiodinase in humans. Thyroid 15, 875–881 (2005).
Makita, N., Miyakawa, M., Fujita, T. & Iiri, T. Sunitinib induces hypothyroidism with a markedly reduced vascularity. Thyroid 20, 323–326 (2010).
Mannavola, D. et al. A novel tyrosine-kinase selective inhibitor, sunitinib, induces transient hypothyroidism by blocking iodine uptake. J. Clin. Endocrinol. Metab. 92, 3531–3534 (2007).
Braun, D. et al. Tyrosine kinase inhibitors noncompetitively inhibit MCT8-mediated iodothyronine transport. J. Clin. Endocrinol. Metab. 97, E100–E105 (2012).
Garfield, D., Hercbergs, A. & Davis, P. Unanswered questions regarding the management of sunitinib-induced hypothyroidism. Nat. Clin. Pract. Oncol. 4, 674 (2007).
Huillard, O. et al. Ocular adverse events of molecularly targeted agents approved in solid tumours: a systematic review. Eur. J. Cancer 50, 638–648 (2014).
Renouf, D. J. et al. Ocular toxicity of targeted therapies. J. Clin. Oncol. 30, 3277–3286 (2012).
Davis, C. Drugs, cancer and end-of-life care: a case study of pharmaceuticalization? Soc. Sci. Med. 131, 207–214 (2015).
Rosen, A. C. et al. Impact of dermatologic adverse events on quality of life in 283 cancer patients: a questionnaire study in a dermatology referral clinic. Am. J. Clin. Dermatol. 14, 327–333 (2013).
Abernethy, A. P. et al. Validation of the Patient Care Monitor (Version 2.0): a review of system assessment instrument for cancer patients. J. Pain Symptom Manage. 40, 545–558 (2010).
Abernethy, A. P. et al. Electronic patient-reported data capture as a foundation of rapid learning cancer care. Med. Care 48, S32–S38 (2010).
Fojo, T. & Parkinson, D. R. Biologically targeted cancer therapy and marginal benefits: are we making too much of too little or are we achieving too little by giving too much? Clin. Cancer Res. 16, 5972–5980 (2010).
Fojo, T. & Grady, C. How much is life worth: cetuximab, non-small cell lung cancer, and the $440 billion question. J. Natl Cancer Inst. 101, 1044–1048 (2009).
Schnipper, L. E., Meropol, N. J. & Brock, D. W. Value and cancer care: toward an equitable future. Clin. Cancer Res. 16, 6004–6008 (2010).
Meropol, N. J. et al. American Society of Clinical Oncology guidance statement: the cost of cancer care. J. Clin. Oncol. 27, 3868–3874 (2009).
Sobrero, A. & Bruzzi, P. Incremental advance or seismic shift? The need to raise the bar of efficacy for drug approval. J. Clin. Oncol. 27, 5868–5873 (2009).
Overbeek, J. A. et al. Costs of hospital events in patients with metastatic colorectal cancer. J. Med. Econ. 14, 656–661 (2011).
Borovicka, J. H. et al. Economic burden of dermatologic adverse events induced by molecularly targeted cancer agents. Arch. Dermatol. 147, 1403–1409 (2011).
Bradbury, P. A. et al. Economic analysis: randomized placebo-controlled clinical trial of erlotinib in advanced non-small cell lung cancer. J. Natl Cancer Inst. 102, 298–306 (2010).
Wong, A. S. et al. Targeted therapy at the end of life for patients with lung cancer. J. Palliat. Med. 13, 945–948 (2010).
Saito, A. M. et al. The effect on survival of continuing chemotherapy to near death. BMC Palliat. Care 10, 14 (2011).
Earle, C. C. et al. Aggressiveness of cancer care near the end of life: is it a quality-of-care issue? J. Clin. Oncol. 26, 3860–3866 (2008).
Soh, T. I. et al. Targeted therapy at the end of life in advanced cancer patients. J. Palliat. Med. 15, 991–997 (2012).
Hui, D. et al. Targeted agent use in cancer patients at the end of life. J. Pain Symptom Manage. 46, 1–8 (2013).
Mintzer, D. M. & Zagrabbe, K. On how increasing numbers of newer cancer therapies further delay referral to hospice: the increasing palliative care imperative. Am. J. Hosp. Palliat. Care 24, 126–130 (2007).
Connor, S. R. U.S. hospice benefits. J. Pain Symptom Manage. 38, 105–109 (2009).
Aldridge Carlson, M. D. et al. Hospices' enrollment policies may contribute to underuse of hospice care in the United States. Health Aff. (Millwood) 31, 2690–2698 (2012).
Cassaratt, D. J. et al. The terrible choice: re-evaluating hospice eligibility criteria for cancer. J. Clin. Oncol. 27, 953–959 (2008).
Kumar, P. & Temel, J. S. End-of-life care discussions in patients with advanced cancer. J. Clin. Oncol. 31, 3315–3319 (2013).
Wright, A. A. et al. Place of death: correlations with quality of life of patients with cancer and predictors of bereaved caregivers' mental health. J. Clin. Oncol. 28, 4457–4464 (2010).
Aldridge, M. D., Canavan, M., Cherlin, E. & Bradley, E. H. Has hospice use changed? 2000–2010 utilization patterns. Med. Care 53, 95–101 (2015).
Teno, J. M. et al. Change in end-of-life care for Medicare beneficiaries: site of death, place of care, and health care transitions in 2000, 2005, and 2009. JAMA 309, 470–477 (2013).
Obermeyer, Z. et al. Association between the Medicare hospice benefit and health care utilization and costs for patients with poor-prognosis cancer. JAMA 312, 1888–1896 (2014).
Rosell, R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 13, 239–246 (2012).
Solomon, B. J. et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 371, 2167–2177 (2014).
Food and Drug Administration. Gilotrif (afatinib): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Inlyta (axitinib): full prescribing information. Drugs@FDA [online], (2012).
Food and Drug Administration. Bosulif (bosutinib): full prescribing information. Drugs@FDA [online], (2012).
Food and Drug Administration. Cometriq (cabozantinib): full prescribing information. Drugs@FDA [online], (2012).
Food and Drug Administration. Zykadia (ceritinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Xalkori (crizotinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Tafinlar (dabrafenib): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Sprycel (dasatinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Tarceva (erlotinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Iressa (gefitinib): drug label. Drugs@FDA [online], (2005).
Food and Drug Administration. Imbruvica (ibrutinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Zydelic (idelalisib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Gleevec (imatinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Tykerb (lapatinib): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Lenvima (lenvatinib): full prescribing information. Drugs@FDA [online], (2015).
Food and Drug Administration. Tasigna (nilotinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Ibrance (palbociclib): full prescribing information. Drugs@FDA [online], (2015).
Food and Drug Administration. Votrient (pazopanib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Iclusig (ponatinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Stivarga (regorafenib): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Jakafi (ruxolitinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Nexavar (sorafenib): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Sutent (sunitinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Mekinist (trametinib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Caprelsa (vandetanib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Zelboraf (vemurafenib): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Kadcyla (ado-trastuzumab emtansine): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Lemtrada (alemtuzumab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Avastin (bevacizumab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Blincyto (blinatumomab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Adcetris (brentuximab vedotin): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Erbitux (cetuximab): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Xgeva (denosumab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Zevalin (ibritumomab tiuxetan): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Yervoy (ipilimumab): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Opdivo (nivolumab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Gazyva (obinutuzumab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Arzerra (ofatumumab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Vectibix (panitumumab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Perjeta (pertuzumab): full prescribing information. Drugs@FDA [online], (2013).
Food and Drug Administration. Cyramza (ramucirumab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Rituxan (rituximab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Sylvant (siltuximab): full prescribing information. Drugs@FDA [online], (2014).
Food and Drug Administration. Bexxar (tositumomab): full prescribing information. Drugs@FDA [online], (2012).
Food and Drug Administration. Herceptin (trastuzumab): full prescribing information. Drugs@FDA [online], (2014).
Nagore, E., Insa, A. & Sanmartin, O. Antineoplastic therapy-induced palmar plantar erythrodysesthesia ('hand-foot') syndrome: incidence, recognition and management. Am. J. Clin. Dermatol. 1, 225–234 (2000).
Acknowledgements
No funding agency in the public, commercial or not-for-profit sectors was used for the preparation of this article. The findings of this work do not necessarily represent the opinion of the US Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
FURTHER INFORMATION
PowerPoint slides
Rights and permissions
About this article

Cite this article
Gharwan, H., Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat Rev Clin Oncol 13, 209–227 (2016). https://doi.org/10.1038/nrclinonc.2015.213
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrclinonc.2015.213
This article is cited by
-
PSMD4 drives progression of hepatocellular carcinoma via Akt/COX2 pathway and p53 inhibition
Human Cell (2023)
-
Proteolysis-targeting chimeras (PROTACs) in cancer therapy
Molecular Cancer (2022)
-
MiR-450a-5p strengthens the drug sensitivity of gefitinib in glioma chemotherapy via regulating autophagy by targeting EGFR
Oncogene (2020)
-
Deciphering novel chemotherapy and its impact on dentistry
British Dental Journal (2020)
