
Abstract
Addictions are prevalent psychiatric disorders that confer remarkable personal and social burden. Despite substantial evidence for their moderate, yet robust, heritability (approx. 50%), specific genetic mechanisms underlying their development and maintenance remain unclear. The goal of this selective review is to highlight progress in unveiling the genetic underpinnings of addiction. First, we revisit the basis for heritable variation in addiction before reviewing the most replicable candidate gene findings and emerging signals from genomewide association studies for alcohol, nicotine and cannabis addictions. Second, we survey the modest but growing field of neurogenetics examining how genetic variation influences corticostriatal structure, function, and connectivity to identify neural mechanisms that may underlie associations between genetic variation and addiction. Third, we outline how extant genomic findings are being used to develop and refine pharmacotherapies. Finally, as sample sizes for genetically informed studies of addiction approach critical mass, we posit five exciting possibilities that may propel further discovery (improved phenotyping, rare variant discovery, gene-environment interplay, epigenetics, and novel neuroimaging designs).
Similar content being viewed by others

Introduction
Excessive use and misuse of psychoactive substances is amongst the leading contributors to morbidity and mortality worldwide [1]. Substance use disorders, as they are referred to in the fifth edition of the Diagnostic and Statistical Manual (DSM) [2], index a highly maladaptive and impairing form of substance use. Previously referred to as abuse and dependence, the current definition of substance use disorders requires the endorsement of two or more of 11 diagnostic criteria [3••].
For the purposes of this review, we refer to substance use disorder as addiction. This term will encompass diagnostic definitions derived from the DSM as well as other diagnostic schema, such as the International Classification of Diseases (e.g., ICD-10) [4] and the Fagerström measures of nicotine dependence (e.g., FTND, FTQ) [5]. The overwhelming majority of the research presented in this review comes from genetic association studies of alcohol, tobacco, and cannabis, with brief mention of emerging research on cocaine and opioids.
Heritability of Addiction
Familial aggregation of addiction has been recognized for decades. Rates of alcoholism in male and female siblings of alcoholic probands have been noted to be 50% and 24% respectively, compared with 20% (male) and 6% (female) of participants from the general population [6]. Rates of other addictions (cannabis, cocaine) and habitual smoking are also elevated, not only in alcoholic probands but also their first-degree relatives. Twin studies have revealed that genetic influences are primarily responsible for this familial aggregation of addiction (see [7] for a review). Regardless of substance, 50% of the variance in addictions can be attributed to those segregating genes that are shared by twin pairs [8]. Notably, such estimates are influenced by a variety of factors, including age and exposure. For example, heritability estimates of substance-related problems are as low as 0% during adolescence, and increase during adolescence and adulthood [9]. This has led to the broad consensus that while familial environment is more important for the early stages of experimentation and substance use, genetic factors exert a more pivotal role in later stages of addiction [10, 11]. Twin studies also substantiate the role of common genetic underpinnings to a variety of addictions, including those involving a host of illicit drugs, alcohol, nicotine, and even caffeine (e.g., [12]). This genetic comorbidity extends beyond addictions to other externalizing problem behaviors as well, including general disinhibitory behavior (e.g., [13•]).
Gene-Finding Efforts
Despite this moderate (50%), yet robust, estimate of heritability and evidence for genetic co-aggregation, gene identification for addiction has been extremely challenging. A majority of these gene-finding efforts have relied on candidates with a priori biological support. Broadly, these genes can be characterized as those participating in the experience of psychoactive effects of the substance (i.e., genes encoding proteins affecting neurotransmitter signaling) and those regulating their bio-availability and clearance (e.g., metabolizing genes), as shown in Fig. 1.

Broad groups of genetic factors influencing addiction and genes that influences this process for alcohol, nicotine and cannabis addiction
Alcohol
Putative Psychoactive Effects (GABRA2).
In human association studies, variation in gamma amino butyric acid receptor A (GABAA) genes, particularly the gene encoding the alpha 2 subunit (GABRA2), have been routinely implicated in the etiology of alcohol addiction [14–18]; however see [19, 20] for examples of non-replications. A recent meta-analysis reports that this association with alcohol dependence is significant for rs279858 (odds-ratio 1.18, p = 5x10-6), a synonymous and frequently studied SNP [21•]. As the primary inhibitory neurotransmitter, GABAergic mechanisms putatively mediate many of the behavioral effects of alcohol, including sedation, motor impairment and withdrawal (see [22] for review). However, GABAA receptors are heteropentameric and receptors containing the α2 subunit are not directly modulated by ethanol [23]. This led investigators to posit that the well-documented associations between GABRA2 SNPs and alcoholism might be attributed to its comorbidity with other externalizing (e.g., impulsivity) or internalizing (e.g., anxiety) disorders. Consistent with this speculation, studies have noted that the same GABRA2 SNPs linked to alcoholism also correlate with its earliest developmental precursor, conduct problems [24]. In particular, a recent study found that impulsiveness was a nearly complete mediator of the association between GABRA2 SNPs and lifetime alcohol problems [25].
In contrast, animal studies assert a more direct pathway between alcohol use and α2 subunit-bearing GABAA receptors. Deletion of the α2 subunit via systemic gene knock-out increased the sedative (and ataxic) effects of ethanol in one study [26] but decreased sedation [27] in another, while altering self-administration rates in neither. In contrast, selective silencing of the gene in the central amygdala temporarily ameliorated binge drinking in rodents [28]. The effects of GABRA2 variants on sedation was recently observed in humans, where those carrying a haplotype spanning 5’ GABRG1 to 3’GABRA2 reported decreased sedation upon challenge with alcohol [29].
Despite this evidence, the precise function of GABRA2 in general, and more specifically, variants studied in humans, remains elusive. The AA genotype (associated with alcoholism) of rs279858 is associated with greater GABRA2 mRNA expression in the prefrontal cortex; however, these mRNA levels did not differ across postmortem brains from alcohol dependent and non-dependent donors [30]. Further, studies imply that the association signal for GABRA2 may actually arise from the neighboring GABRG1 gene [31, 32]. Both GABRA2 and GABRG1 expression appear to be down-regulated in the hippocampus of alcohol dependent individuals [33]. As γ1-subunit containing GABAA receptors are only expressed in selected brain regions (e.g., central amygdala) that have been implicated in addiction, they have emerged as possible therapeutic targets [34]. However, the extent to which these receptors respond to ethanol remains unknown.
Metabolism (ADH1B, ALDH2).
Two polymorphisms in genes encoding alcohol metabolizing enzymes, alcohol dehydrogenase 1B (ADH1B) and aldehyde dehydrogenase 2 (ALDH2) have been associated with differences in alcohol consumption and dependence [35–37]. The ADH1B Arg48His (rs1229984), results in increased oxidation of alcohol to acetaldehyde, while the other variant, ALDH2 Glu504Lys (rs671), is associated with decreased enzymatic conversion of acetaldehyde to acetate. Consequently both variants, most prevalent among East Asians [38], cause the accumulation of acetaldehyde, which induces aversive reactions to alcohol consumption including flushing, nausea, and palpitations. This aversive response typically decreases alcohol consumption and protects against the development of addiction. Although relatively uncommon in non-Asians, the protective effects of the ADH1B allele on alcohol dependence and consumption were recently supported within a meta-analysis of individuals of European and African American descent [39] and more recently, at genomewide significant levels in a German population [40] and in an independent genomewide association study of European and African-American subjects [41••]. In addition to ADH1B and ALDH2, other variants in alcohol dehydrogenase encoding genes, such as ADH1A, ADH1C, ADH4, ADH5, and ADH7 have also been sporadically associated with alcohol dependence [36, 37, 42–44].
Nicotine
Putative Psychoactive Effect (CHRNA5-A3-B4).
Among the most replicated associations in addiction are those between polymorphisms in the nicotinic acetylcholine receptor gene cluster encoding the α5, α3, and β4 subunits (CHRNA5-A3-B4) with nicotine dependence [45–47], smoking behaviors [48, 49], and treatment response [50]. A majority of studies implicate a non-synonymous SNP, rs16969968, in CHRNA5 encoding an aspartic acid to asparagine substitution at codon 398 (a highly conserved residue), and a highly correlated proxy SNP, rs1051730, in CHRNA3. This variant (rs16969968) was initially identified in a candidate gene association study of nicotine dependent cases [45]. A proliferation of studies followed when multiple genomewide association studies unequivocally identified this variant as a contributor to genetic vulnerability for cigarettes smoked per day (CPD). Aggregating effect sizes across 73,853 subjects, investigators convincingly documented an association on the order of 10-73 for an association between rs1051730 and CPD [51, 52, 53••].
The intuitive appeal of the signal coming from CHRNA3/CHRNA5 arises from the putative effects of rs16969968 on receptor function (but not expression). As α4β2α5 receptors with the asparagine residue show decreased response to a nicotine agonist, carriers may require greater amounts of nicotine and thus, be more vulnerable to dependence [47] (but see also [54]). In addition, α5 knock-out mice appear to be more sensitive to the rewarding effects of nicotine, particularly at higher concentrations, demonstrating profound increases in motivation to seek and consume nicotine [55].
In addition to rs16969968, several other variants in this gene cluster (e.g., rs680244, associated with gene expression) have been associated with smoking as well as alcohol use. Broadly speaking, the linkage disequilibrium (LD) patterns across the genes indicate the presence of 3-4 bins tagged by variants - rs3841324 (CHRNA5 promoter), rs16969968, and rs578776 (3’-UTR of CHRNA3) [56] and there is evidence that these LD bins vary in their relationships with smoking, across populations [57, 58], with each exerting putatively independent effects.
Metabolism (CYP2A6).
Nicotine is primarily metabolized by a liver enzyme, cytochrome P450 2A6 (CYP2A6), which converts nicotine to cotinine (COT), and further to 3’-hydroxycotinine (3HC) [59, 60]. The ratio of 3HC/COT is a robust and highly heritable (47-67%) marker of nicotine clearance and of metabolic activity [61] and polymorphisms in the CYP2A6 gene explain 6-19% of this heritable variation [62]. There are numerous polymorphisms in CYP2A6 and their activities range from fully inactive alleles (e.g., CYP2A6*2) to those that reduce enzymatic efficiency by varying degrees (e.g., CYP2A6*9) [63]. Based on their activity, CYP2A6 alleles have been used to classify individuals into slow (homozygotes/carriers for no activity allele or carriers for reduced activity allele), intermediate (carriers for reduced activity allele) and normal metabolizers [64]. These correspond, roughly, to <50%, 50-80% and >80% CYP2A6 activity. Individuals with CYP2A6 genotypes conferring slower metabolism tend to report smoking fewer CPD, less nicotine dependence, and are more likely to quit by themselves [65, 66] (except see [67]) or with pharmacologic treatment [68] (see also [69]). However, relative to variants in other genes, including CHRNA5/CHRNA3, CYP2A6 associations less robustly predict nicotine addiction. For example, a genomewide association study (GWAS) meta-analysis found an association between a variant (rs4105144) in linkage disequilibrium with a metabolically deficient CYP2A6*2 allele (and other reduced activity alleles) and daily cigarette consumption, but not nicotine dependence [52].
Cannabis
Putative Psychoactive Effects (CNR1).
CNR1 encodes the human endocannabinoid receptor 1 (CB1) which is the putative binding site for Δ9-tetrahydrocannabinol. CB1 knock-out mice do not display effects associated with cannabinoids (e.g., antinociception) [70]. Numerous studies have examined the relationship between variants in CNR1 and cannabis addiction as well as other substance use disorders (for review see [71]). The most frequently studied of these polymorphisms is the (AAT)n repeat in the 3’ untranslated region of the gene, a variable number of tandem repeats with unknown functional consequence. A recent meta-analysis reported that individuals with greater than 15 repeats may be at increased risk for addiction to illicit drugs [72•]. Other variants (e.g., rs1049353, rs806380) have produced inconsistent results, further complicated by the examination in some studies of addiction to all illicit drugs (even though cannabis is likely to be the most commonly used and misused of these).
Metabolism (CYP2C9).
Cannabis is first converted to an active metabolite, 11-hydroxy-delta-9-tetrahydrocannabinol (11-OH-THC), and then to its inactive form, 11-nor-9-carboxy-delta-9-tetrahydrocannabinol (THC-COOH) by the cytochrome family of enzymes [73, 74]. We are only aware of one study that found that CYP2C9*3 carriers were more likely to have higher concentrations of THC and, correspondingly, lower concentrations of THC-COOH and show greater sedation upon oral THC administration [75].
GWAS and Identification of Novel Variants
GWAS, when well powered, can serve as a powerful tool for uncovering common variation associated with addiction phenotypes. For instance, in the largest of the aforementioned meta-analyses of CPD, an aggregate sample size > 140,000 resulted in an association signal for rs1051730 in CHRNA3 on the order of magnitude of 10-73 [53••]. Despite the high degree of statistical significance, the effect size was small with each risk allele only associated with one extra CPD. Such weak penetrance likely limits the ability of individual GWAS to produce replication. There have now been nine GWAS on DSM-IV alcohol dependence [76–83] and of these, only three report genomewide significant signals. The first two report signals in PECR (Peroxisomal Trans-2-Enoyl-CoA Reductase) [82] and ADH1C (alcohol dehydrogenase 1C) [83], however none of the other studies have replicated these associations. The most recent of these is perhaps the most promising, convincingly demonstrating association between a symptom count of alcohol dependence and SNPs in ADH1B, including rs1229984 (p = 1.2E-31) in European- and rs2066702 (p = 6.3E-17) in African-American subjects [41••]. One meta-analysis has also identified a significant association between daily typical alcohol intake and a variant (rs6943555) within the autism susceptibility candidate 2 (AUTS2) gene (P = 4.1E-9) [84].
While meta-analyses are pending for other substances, individual GWAS have yielded promising leads for cocaine and opioids. A recent GWAS of cocaine dependence identified rs2629540 in FAM53B [85]. The same group of investigators has also recently reported genomewide significant association signals for genes in potassium signalling pathways for opioid dependence [86]. In contrast, results for cannabis use and dependence have been less promising [87, 88]. Despite the lack of statistically significant findings, there is growing evidence that common GWAS variation explains 24-38% of the variation in current smoking [89] and alcoholism [90].
Neurogenetic Developments
A significant challenge of addiction genetics research involves ascribing function to individual polymorphisms and isolating their role in the addictive process. Evaluating the functional mechanisms of polymorphisms associated with the psychoactive effects of substances is further complicated by the fact that they likely involve neural intermediates. We focus here on neuroimaging outcomes as a promising complement to the association studies discussed above.
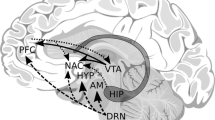
The corticostriatal system, which includes the striatum, ventromedial prefrontal cortex (vmPFC), orbitofrontal cortex (OFC), thalamus, insula, hippocampus, and anterior cingulate cortex (ACC), among other structures, is critical for reward processing (for review see [91•]). Emerging neurogenetics research [92•] suggests that genetic variation associated with addiction risk also predicts individual differences in corticostriatal function, structure, and connectivity, providing putative mediating neural mechanisms through which these polymorphisms promote addictions. While the vast majority of addiction-related neurogenetics research has focused on genetic variation in the dopaminergic system [93, 94], we focus our review on research evaluating variation within GABRA2, CHRNA5-CHRNA3-CHRNB4, and CNR1.
Alcohol and GABRA2.
Two studies have found that genotypes predictive of alcohol addiction (i.e., rs279871 A homozygotes, rs279826 G homozygotes, rs279858 G homozygotes), are also associated with enhanced neural response to reward predictors in brain regions associated with subjective valuation, including the vmPFC [95] and insula [96••]. While these studies suggest that GABRA2 risk variants may confer vulnerability to alcohol addiction by enhancing the subjective valuation of external cues, there was no evidence that genotype predicted craving [95]. One possibility is that this enhanced reactivity in regions critical for subjective valuation may drive urgent and impulsive attempts to regulate negative emotional states [96••].
Nicotine and CHRNA5-A3-B4.
Risk-related polymorphisms in CHRNA5-A3-B4 have been associated with individual differences in corticostriatal circuit reactivity to nicotine cues as well as resting state connectivity. The risk (i.e., A) allele of rs16969968 predicts reduced reactivity in corticostriatal circuit nodes (e.g., dorsal striatum, hippocampus, insula) in response to smoking cues among nicotine-dependent women [97]. Notably, because smoking is associated with enhanced smoking cue reactivity, these results suggest a dissociation between CHRNA5-A3-B4 genetic risk and neural correlates of smoking. While the reasons for this dissociation are unclear, the authors speculate that risk allele carriers may form relatively poorer drug-cue associations and that stronger cue-related memory may be needed to promote continued smoking in those with relatively lower genetic risk. Another possible interpretation of these data is that because the risk allele at rs16969968 is associated with decreased response to a nicotine agonist [47], independent smoking-related cues may not develop the same predictive value for risk allele carriers, who may need to ingest relatively larger dosages to obtain reward. The risk allele of rs16969968 may confer vulnerability to smoking via blunted corticostriatal response to smoking cues, or via other, not mutually exclusive neural mechanisms, such as neural reactivity to threat [98].
The only other neuroimaging study we are aware of investigating CHRNA5-A3-B4, studied resting-state functional connectivity (i.e., the synchronization of intrinsic low-frequency fluctuations between brain regions outside of task performance) within the corticostriatal circuit. Similar to smokers [99], rs16969968 risk allele carriers had reduced functional coupling of the dorsal anterior cingulate cortex (dACC) and striatum/extended amygdala, regardless of smoking status [100]. This study also found that CHRNA3 rs578776 G (risk) allele carriers had elevated dACC and thalamic resting state connectivity, which was correlated with smoking just prior to the imaging session (measured via expired CO levels). Given these differential associations with lifetime and immediate smoking, and the extant literature demonstrating the independence of their effects, the authors posit that rs16969968 and rs578776 genotypes contribute to trait and state aspects of smoking respectively [100]. In contrast to the study by Janes and colleagues [97], the results of this study suggest that CHRNA5-A3-B4 genetic risk covaries with corticostriatal correlates of smoking.
Cannabis and CNR1.
Genetic variation in the endocannabinoid system has been implicated in neural response during abstinence-related withdrawal and craving [101–103]. Specifically, during abstinence, rs2023239 (non-synonymous) heterozygotes have elevated reactivity to marijuana cues in OFC and ACC relative to A allele homozygotes [103]. This elevated reactivity in corticostriatal regions critical for reward processing and the valuation of external stimuli [102] may contribute to increased marijuana craving in rs2023239 heterozygotes following abstinence [101]. Notably, a functional variant (rs324420) in FAAH, which codes for the fatty acid amide hydrolase enzyme that catabolizes endocannabinoids, was found to have independent and additive effects along with CNR1 [103••]. In addition to mediating neural activity in response to cues, CNR1 variants have also been linked to altered brain morphology. For example, Schacht et al. [104] found that heavy cannabis users have lower bilateral hippocampal volume than healthy controls, with the smallest volumes found in cannabis users with the rs2023239 G allele. Given that the hippocampus is a region critical in memory processing, these findings provide a putative mechanism underlying the association between heavy cannabis use and deficits in cognitive processes and learning [105].
The few neurogenetic studies of GABRA2, CHRNA5-CHRNA3-CHRNB4, and CNR1 genotypes associated with addiction risk suggest that these polymorphisms may drive addiction risk via their effects on corticostriatal connectivity and reactivity to reward. Notably, while we concentrated on neurogenetic research investigating genetic polymorphisms that are believed to moderate the psychoactive effects of substances, new research suggests that this approach may also afford a remarkable opportunity to examine whether indices of metabolism exert an impact on neural systems. For instance, a revealing new study [106] found that normal metabolizers (defined by CYP2A6 genotype) have greater smoking cue-induced activation in the right amygdala, striatum, hippocampus, and cingulate cortex relative to reduced metabolizers. Such studies of metabolic-related polymorphisms may be particularly informative in the context of drug-challenge paradigms.
Putting your Genes to work: Pharmacogenetics
Given the contrast between the modest effect sizes attributable to individual variants and the relatively large impact of environmental control that can be exerted (e.g., increasing taxation, prohibition of use in public places), are genetic studies of addiction worthwhile [107]? Perhaps the most compelling response to this question comes from emerging pharmacogenetic efforts that attempt to tailor pharmacotherapy based on genotype.
Alcohol.
In the broadest sense, disulfuram (Antabuse), which inhibits aldehyde dehydrogenase (responsible for conversion of acetaldehyde to acetate) and targets the action of ALDH2 might be considered a drug with pharmacogenetic underpinnings. Although non-Asians do not carry the protective variant of rs671, disulfuram approximates its effects by inducing aversive reactions upon alcohol intake. A more specific instance in which genotype modulates treatment response for alcoholism comes from well-replicated studies of the Asn40Asp variant (rs1799971) in OPRM1 which encodes the mu-opioid receptor, and moderates the effectiveness of naltrexone (NTX; a mu-opioid antagonist) treatment [108]. Recent findings show that Asp40 (G) allele carriers have more positive subjective responses [109] and increased mesolimbic dopamine release following alcohol challenge compared to those homozygous for the Asn40 allele [A] [110]. There is evidence that NTX is efficacious in reducing heavy drinking only in Asp40 carriers [111–113]. GABRA2 variants also have been found to moderate treatment outcomes. Specifically, rs279858 was associated with drinking in individuals assigned to different psychotherapeutic treatments [114]. Although in that study there were no appreciable differences across treatment type (12-step vs. cognitive behavioral vs. motivational enhancement therapy), individuals homozygous for the low-risk A allele, had fewer drinking days when enrolled in 12-step treatment. In another study, carriers of the same risk allele relapsed to drinking earlier than A-allele homozygotes [115].
Another intriguing series of experiments have focused on tailoring medications for alcoholism that act via serotonergic modulation. Two drugs, ondansetron and sertraline, are worth mention for their differential efficacy in the context of individual genotype (short S versus long L) at the promoter polymorphic region (5-HTTLPR) of the serotonin transporter (SLC6A4; 5-HTT) gene. Ondansetron is an antagonist of the serotonin (5-HT3) receptors. One study found that the most substantial reduction in drinks/day and days abstinent upon treatment was noted in those who were homozygous for the long (LL) allele of 5-HTTLPR and the T allele of a 3’ untranslated region (UTR) variant, rs1042173, putatively due to increased drug-provoked blockade of upregulated post-synaptic serotonin receptors in LL/TT individuals [116, 117]. Sertraline, on the other hand, is a selective serotonin reuptake inhibitor (SSRI) and has been found to be more efficacious, in general, in those with comorbid depression [118]. These observations led to the finding that while sertraline was beneficial in LL individuals who were late-onset alcoholics (typically comorbid with internalizing features [118]), it performed worse than placebo in early-onset cases [119], particularly in the context of daily anxiety.
Nicotine.
Both CHRNA5-CHRNA3-CHRNB4 and CYP2A6 have been implicated in treatment response. For example, the effectiveness of nicotine replacement therapy (but not bupropion) has been found to be more pronounced in fast versus slow metabolizers [69]. A study has also shown that slow metabolizers tend to require fewer doses of nicotine spray but do not differ in their response to patch usage [120], although there is emerging evidence that slow metabolizers may be at higher risk for patch-related nicotine toxicity [121]. Similarly, one study showed that a high-risk haplotype comprised of variants in CHRNA5-CHRNA3-CHRNB4 predicted failed abstinence in placebo-treated subjects but not those receiving active pharmacotherapy [50], while another study reported similar findings for the placebo group but greater abstinence in those receiving nicotine replacement therapy [122]. Perhaps the most prominent of therapeutic alternatives for smoking cessation is varenicline, a partial agonist of the α4β2 nicotinic receptor subtype. Variants in CHRNA4 and CHRNB2 have been implicated in response to varenicline while variants in CHRNB4 were associated with nausea, a common side effect of medication use, which typically attenuates adherence [123].
Opioids.
Opioids, particularly heroin, are drugs with extremely high addictive potential [124]. Treatments for opioid dependence can vary from the typical use of methadone (opioid agonist) and buprenorphine (partial agonist) to even naltrexone (opioid antagonist). Response rates vary dramatically and the use of none of these is without risks [125]. One recent study of African-Americans found that for a variant, rs678849, in the gene encoding the delta opioid receptor (OPRD1), CC individuals had more positive urine tests (24 weeks post-treatment) if treated with buprenorphine but less positive tests when treated with methadone [126]. Another recent study implicates a functional variant (rs2760118) in the gene encoding the succinic semialdehyde dehydrogenase enzyme (ALDH5A1) in response to methadone maintenance with T allele carriers more likely to be non-responders [127].
Where do we go from here?
“It was the best of times, it was the worst of times, it was the age of wisdom, it was the age of foolishness…”[128]. These are exciting times for the genetic study of addictions. What is next in this “age of wisdom”? We propose the following as promising avenues for future research:
-
1.
We are what we measure: Early addiction studies relied heavily on diagnostic entities (e.g., alcohol dependence), assuming homogeneity within the construct. Evidence from rodent models (e.g., [129]) substantiated by human genetic studies (e.g., [130•, 131]) imply that such an assumption of homogeneity, particularly for alcohol and nicotine addictions, may be inaccurate. Aspects of addiction that may appear non-diagnostic are also worth study. For instance, cocaine-induced paranoia, a dangerous consequence of excessive cocaine use has been successfully investigated with particular attention to variants in dopamine beta-hydroxylase (DBH; associated with dopamine metabolism) (e.g., [132]). This research complements findings from a recent pharmacogenetic trial that implicated rs1611115 in DBH in predicting efficacy of a novel vaccine for cocaine misuse [133], such that vaccinated carriers of the T allele (putatively associated with lower DβH activity) showed a more dramatic decrease (77 to 51% versus 83 to 72% in CC individuals) in positive urines across a 16-week study period [134]. Sample size restrictions remain an essential barrier to the study of individual components or sequelae of addictions, and may require institutional support for data collection efforts targeted at phenotypic refinement. To this end, another possibility is to investigate those features that underpin a variety of addictions, for instance impulsivity and compulsivity. Such behaviors are frequently assessed across a variety of studies as they integrate not only across substance use behaviors but also likely underpin behavioral addictions (e.g., disordered gambling [135]), including the more recent and controversial notion of food addictions [136].
-
2.
Rare variants for commonly occurring disorders: GWAS capitalize on the common disorder-common variant hypothesis, and as such, this is proving to be fruitful for even rarer psychiatric disorders (e.g., schizophrenia). Nonetheless, existing exome arrays and emerging sequencing strategies present opportunities for the study of rare variants for these commonly occurring addictions [137]. For instance, Vrieze et al. [138] recently reported that even though individual rare variants were not statistically significant, when combined with common (GWAS) variation, these polymorphisms explained up to 84% of the heritable variation in illicit drug use [138]. Another study conducted targeted deep sequencing of 17 ionotropic glutamate (N-methyl-D-aspartate) receptors and identified rare variants in the disrupted in schizophrenia (DISC1; rs139667828) and glutamate receptor, ionotropic, N-methyl D-aspartate 2B (GRIN2B; rs146792012) gene for opioid dependence in African-Americans [139]. The study of rare variants is not without challenges, in particular, the requirement for large samples. Family data is also exceptionally valuable in their study but methodologies for the study of general enrichment or the specific transmission of individual variants are still being honed, particularly for whole genome data.
-
3.
Genes and environment: Even in this era of skepticism [140, 141], there is little doubt that the etiology of addictions involves gene-environment interplay. Even for variants impacting metabolism, such as rs671 in ALDH2, there is evidence that increasing social pressures to drink have contributed to cohort-related increases in alcohol consumption in heterozygous carriers, who drink despite flushing [142]. There is also evidence that childhood adversity intensifies the risk posed by absence of the protective allele of rs1229984 (ADH1B;[143]). Such environmental moderation is likely to be even more pronounced for genes that regulate the psychoactive effects of these substances (e.g., GABRA2 genotypes and parental monitoring,[24]; CHRNA5 SNPs and peer smoking [144], parental monitoring [145], childhood adversity [146], and early age of onset [147]). Thus, if statistical challenges can be surmounted, this is an exceedingly promising avenue for future enquiry.
-
4.
Epigenetic (dys)regulation: The impact of environmental adversity, particularly during childhood, may extend well beyond moderation of changes in DNA sequence to expression differences that are epigenetic in nature (i.e., changes in gene expression unrelated to sequence) [148]. For instance, hypermethylation of CpG sites of genes such as ALDH1A1 and CHRNA5 have been noted in peripheral tissue from alcoholic patients with a history of childhood adversity [149]. Relative to controls, early studies also reported increased global methylation in leukocytes from alcoholic subjects [150], in particular in the promoter regions of genes implicated in alcohol addictions, such as OPRM1 [151] and the dopamine transporter (SLC6A3), with the latter predicting craving severity [152]. The principal challenge with epigenetic studies is the use of peripheral tissue and their uncertain association with regionally specific differences within the brain [153]. Although relatively few studies have investigated epigenetic mechanisms within the post-mortem brain, one study [154] found increased methylation levels in 3 prodynorphin (PDYN) CpG-SNPs in the dorsolateral prefrontal cortex of alcohol dependent subjects.
-
5.
Drugs and the developing brain: A majority of neurogenetics, and in fact, neuroimaging studies have relied on samples of addicted individuals responding to cues. Such cross-sectional studies pose the quintessential dilemma of whether differences noted can be attributed to predisposing characteristics that preceded drug exposure (e.g., family history) or whether they are a consequence of prolonged drug use. Twin pairs discordant for addiction status offer an elegant control for genetic liability. Utilizing this paradigm, one study found that relative to their exposed but non-regular smoking identical co-twin, the genetically identical twin who smoked regularly showed no differences in corticostriatal response to reward when whole brain analyses were conducted, although regions associated with cognitive control were differentially activated [155]. This suggests that extant findings implicating structures in the corticostriatal system in the etiology of addiction relate to inherited differences, rather than variations that evolve as a function of drug exposure. Likewise, prospective neurogenetic studies of addiction are of immediate importance.
Conclusions
Table 1 provides an overview of key accomplishments in addiction genetics. We detail there a handful of key genes in relation to their role in the psychoactive effects and metabolism of alcohol, nicotine, or cannabis. However, there has not been consistent replication of these findings. GWAS are beginning to reveal intriguing findings and meta-analyses that will aggregate data across these individual GWAS have begun to be formulated. However, unlike schizophrenia [156], such efforts lag considerably in accruing sample sizes large enough to detect variants of the expected modest effect size. Neurogenetic studies continue to inform the functional relevance of polymorphisms uncovered using genetic association methods, however they are limited by the phenotypes they investigate (e.g., how does craving relate to general addiction?), how these phenotypes relate to addiction (e.g., how do genes that affect corticostriatal response relate to alcoholism?) and low statistical power. In response to these challenges, it is likely that advances in addictions research over the next decade will stem from collaborative and translational science. There are other exciting avenues for translational research that are beyond the scope of this review, such as the utility of animal models (e.g., rodent [157], and drosophila models [158] of drinking) or the remarkable potential of using skin biopsies from addicted individuals to induce pluripotent stem cells that differentiate into neuronal tissue and examining the effect of ethanol exposure to live tissue [159]. Such modest efforts at examining the many facets of the biological processes underlying addictions will eventually culminate in great strides, both in scientific discovery and in our considerably enhanced ability to treat addictions - common, yet personally and societally burdensome mental disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Degenhardt L, Hall W. Extent of illicit drug use and dependence, and their contribution to the global burden of disease. Lancet. 2012;379(9810):55–70.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: American Psychiatric Association; 2013. Xliv, 947.
Hasin DS et al. DSM-5 criteria for substance use disorders: recommendations and rationale. Am J Psychiatry. 2013;170(8):834–51. This paper, authored by the DSM-5 workgroup, outlines the rationale for the diagnostic changes implemented for substance use disorders.
World Health Organization. The ICD-10 classification of mental and behavioural disorders : clinical descriptions and diagnostic guidelines. Geneva: World Health Organization; 1992. xii, 362 p.
Heatherton TF et al. The Fagerstrom Test for Nicotine Dependence: a revision of the Fagerstrom Tolerance Questionnaire. Br J Addict. 1991;86(9):1119–27.
Bierut LJ et al. Familial transmission of substance dependence: alcohol, marijuana, cocaine, and habitual smoking: a report from the Collaborative Study on the Genetics of Alcoholism. Arch Gen Psychiatry. 1998;55(11):982–8.
Agrawal A, Lynskey MT. Are there genetic influences on addiction: evidence from family, adoption and twin studies. Addiction. 2008;103(7):1069–81.
Agrawal A et al. The genetics of addiction-a translational perspective. Transl Psychiatry. 2012;2:e140.
Kendler KS et al. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry. 2008;65(6):674–82.
Baker JH, Maes HH, Kendler KS. Shared environmental contributions to substance use. Behav Genet. 2012;42(3):345–53.
Rhee SH et al. Genetic and environmental influences on substance initiation, use, and problem use in adolescents. Arch Gen Psychiatry. 2003;60(12):1256–64.
Kendler KS, Myers J, Prescott CA. Specificity of genetic and environmental risk factors for symptoms of cannabis, cocaine, alcohol, caffeine, and nicotine dependence. Arch Gen Psychiatry. 2007;64(11):1313–20.
Hicks BM et al. Psychometric and genetic architecture of substance use disorder and behavioral disinhibition measures for gene association studies. Behav Genet. 2011;41(4):459–75. Shows that underlying substance use, substance use disorders and disinhibition are shared genetic influences.
Edenberg HJ et al. Variations in GABRA2, encoding the alpha 2 subunit of the GABA(A) receptor, are associated with alcohol dependence and with brain oscillations. Am J Hum Genet. 2004;74(4):705–14.
Covault J et al. Allelic and haplotypic association of GABRA2 with alcohol dependence. Am J Med Genet B Neuropsychiatr Genet. 2004;129B(1):104–9.
Lappalainen J et al. Association Between Alcoholism and γ-Amino Butyric Acid α2 Receptor Subtype in a Russian Population. Alcohol: Clin Exp Res. 2005;29(4):493–8.
Fehr C et al. Confirmation of association of the GABRA2 gene with alcohol dependence by subtype-specific analysis. Psychiatr Genet. 2006;16(1):9–17.
Soyka M et al. GABA-A2 receptor subunit gene (GABRA2) polymorphisms and risk for alcohol dependence. J Psychiatr Res. 2008;42(3):184–91.
Lind PA et al. The role of GABRA2 in alcohol dependence, smoking, and illicit drug use in an Australian population sample. Alcohol Clin Exp Res. 2008;32(10):1721–31.
Onori N et al. GABRA2 and alcohol use disorders: no evidence of an association in an Italian case-control study. Alcohol Clin Exp Res. 2010;34(4):659–68.
Li D et al. Association of Gamma-Aminobutyric Acid A Receptor alpha 2 Gene (GABRA2) with Alcohol Use Disorder. Neuropsychopharmacol. 2013. doi:10.1038/npp.2013.291. Meta-analysis of variants in GABRA2 and their relationship with alcohol use disorders demonstrating robustness of this association.
Kumar S et al. The role of GABA(A) receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacol (Berl). 2009;205(4):529–64.
Paul SM. Alcohol-sensitive GABA receptors and alcohol antagonists. Proc Natl Acad Sci U S A. 2006;103(22):8307–8.
Dick DM et al. Role of GABRA2 in trajectories of externalizing behavior across development and evidence of moderation by parental monitoring. Arch Gen Psychiatry. 2009;66(6):649–57.
Villafuerte S et al. Impulsiveness mediates the association between GABRA2 SNPs and lifetime alcohol problems. Genes Brain Behav. 2013;12(5):525–31.
Dixon CI et al. Deletion of the gabra2 gene results in hypersensitivity to the acute effects of ethanol but does not alter ethanol self administration. PLoS One. 2012;7(10):e47135.
Boehm 2nd SL et al. Gamma-Aminobutyric acid A receptor subunit mutant mice: new perspectives on alcohol actions. Biochem Pharmacol. 2004;68(8):1581–602.
Liu J et al. Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci U S A. 2011;108(11):4465–70.
Uhart M et al. GABRA2 markers moderate the subjective effects of alcohol. Addict Biol. 2013;18(2):357–69.
Haughey HM et al. Human gamma-aminobutyric acid A receptor alpha2 gene moderates the acute effects of alcohol and brain mRNA expression. Genes Brain Behav. 2008;7(4):447–54.
Covault J et al. Markers in the 5'-region of GABRG1 associate to alcohol dependence and are in linkage disequilibrium with markers in the adjacent GABRA2 gene. Neuropsychopharmacology. 2008;33(4):837–48.
Ittiwut C et al. GABRG1 and GABRA2 variation associated with alcohol dependence in African Americans. Alcohol Clin Exp Res. 2012;36(4):588–93.
Enoch MA et al. GABAergic gene expression in postmortem hippocampus from alcoholics and cocaine addicts; corresponding findings in alcohol-naive P and NP rats. PLoS One. 2012;7(1):e29369.
Khom S et al. Pharmacological properties of GABAA receptors containing gamma1 subunits. Mol Pharmacol. 2006;69(2):640–9.
Li D, Zhao H, Gelernter J. Strong association of the Alcohol Dehydrogenase 1B Gene (ADH1B) with alcohol dependence and alcohol-induced medical diseases. Biol Psychiatry. 2011;70(6):504–12.
Luo X et al. Diplotype trend regression analysis of the ADH gene cluster and the ALDH2 Gene: multiple significant associations with alcohol dependence. Am J Hum Genet. 2006;78(6):973–87.
Macgregor S et al. Associations of ADH and ALDH2 gene variation with self report alcohol reactions, consumption and dependence: an integrated analysis. Hum Mol Genet. 2009;18(3):580–93.
Goedde HW et al. Distribution of ADH2 and ALDH2 genotypes in different populations. Hum Genet. 1992;88(3):344–6.
Bierut LJ et al. ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Mol Psychiatry. 2012;17(4):445–50.
Treutlein J, et al. ADH1B Arg48His allele frequency map: filling in the gap for Central Europe. Biol Psychiatry. 2013.
Gelernter J, et al. Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry, 2014;19(1):41–49. First genomewide association study to implicate ADH1B variants in European- and African American subjects at p < 5 x 10 -8 .
Gizer IR et al. Association of alcohol dehydrogenase genes with alcohol-related phenotypes in a Native American community sample. Alcohol Clin Exp Res. 2011;35(11):2008–18.
Edenberg HJ, Foroud T. The genetics of alcoholism: identifying specific genes through family studies. Addict Biol. 2006;11(3–4):386–96.
Edenberg HJ et al. Association of alcohol dehydrogenase genes with alcohol dependence: a comprehensive analysis. Hum Mol Genet. 2006;15(9):1539–49.
Saccone SF et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16(1):36–49.
Weiss RB et al. A candidate gene approach identifies the CHRNA5–A3-B4 region as a risk factor for age-dependent nicotine addiction. Plos Genetics. 2008;4(7):e1000125.
Bierut LJ et al. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008;165(9):1163–71.
Ducci F et al. TTC12-ANKK1-DRD2 and CHRNA5-CHRNA3-CHRNB4 influence different pathways leading to smoking behavior from adolescence to mid-adulthood. Biol Psychiatry. 2011;69(7):650–60.
Ware JJ, van den Bree MB, Munafo MR. Association of the CHRNA5-A3-B4 gene cluster with heaviness of smoking: a meta-analysis. Nicotine Tob Res. 2011;13(12):1167–75.
Chen LS et al. Interplay of genetic risk factors (CHRNA5-CHRNA3-CHRNB4) and cessation treatments in smoking cessation success. Am J Psychiatry. 2012;169(7):735–42.
Liu JZ et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42(5):436–40.
Thorgeirsson TE et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42(5):448–53.
Tobacco and C. Genetics, Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet, 2010. 42(5): p. 441-7. Meta-analysis of multiple large samples implicating the role of rs1051730, which is highly correlated with a functional variant, rs16969968, for its association with cigarettes smoked per day.
Jackson KJ et al. Role of alpha5 nicotinic acetylcholine receptors in pharmacological and behavioral effects of nicotine in mice. J Pharmacol Exp Ther. 2010;334(1):137–46.
Fowler CD et al. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471(7340):597–601.
Wang JC et al. Genetic variation in the CHRNA5 gene affects mRNA levels and is associated with risk for alcohol dependence. Mol Psychiatry. 2009;14(5):501–10.
Chen LS et al. Smoking and genetic risk variation across populations of European, Asian, and African American ancestry–a meta-analysis of chromosome 15q25. Genet Epidemiol. 2012;36(4):340–51.
Saccone NL et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS Genet. 2010;6(8):e1001053.
Hukkanen J, Jacob 3rd P, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57(1):79–115.
Yamazaki H et al. Roles of CYP2A6 and CYP2B6 in nicotine C-oxidation by human liver microsomes. Arch Toxicol. 1999;73(2):65–70.
Dempsey D et al. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther. 2004;76(1):64–72.
Swan GE et al. Genetic and environmental influences on the ratio of 3'hydroxycotinine to cotinine in plasma and urine. Pharmacogenet Genomics. 2009;19(5):388–98.
Bloom AJ et al. Use of a predictive model derived from in vivo endophenotype measurements to demonstrate associations with a complex locus, CYP2A6. Hum Mol Genet. 2012;21(13):3050–62.
Ray R, Tyndale RF, Lerman C. Nicotine dependence pharmacogenetics: role of genetic variation in nicotine-metabolizing enzymes. J Neurogenet. 2009;23(3):252–61.
Chenoweth MJ et al. CYP2A6 slow nicotine metabolism is associated with increased quitting by adolescent smokers. Pharmacogenet Genomics. 2013;23(4):232–5.
Schoedel KA et al. Ethnic variation in CYP2A6 and association of genetically slow nicotine metabolism and smoking in adult Caucasians. Pharmacogenetics. 2004;14(9):615–26.
Rubinstein ML et al. Nicotine metabolism and addiction among adolescent smokers. Addiction. 2013;108(2):406–12.
Lerman C et al. Genetic variation in nicotine metabolism predicts the efficacy of extended-duration transdermal nicotine therapy. Clin Pharmacol Ther. 2010;87(5):553–7.
Chen LS, et al. Pharmacotherapy effects on smoking cessation vary with nicotine metabolism gene (CYP2A6). Addiction. 2013.
Ledent C. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283(5400):401–4.
Agrawal A, Lynskey MT. Candidate genes for cannabis use disorders: findings, challenges and directions. Addiction. 2009;104(4):518–32.
Benyamina A et al. CNR1 gene polymorphisms in addictive disorders: a systematic review and a meta-analysis. Addict Biol. 2011;16(1):1–6. Meta-analysis of variants in CNR1 implicating the AAT repeat as being weakly associated with illicit drug addictions.
Watanabe K et al. Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci. 2007;80(15):1415–9.
Huestis MA, Barnes A, Smith ML. Estimating the time of last cannabis use from plasma delta9-tetrahydrocannabinol and 11-nor-9-carboxy-delta9-tetrahydrocannabinol concentrations. Clin Chem. 2005;51(12):2289–95.
Sachse-Seeboth C et al. Interindividual variation in the pharmacokinetics of Delta9-tetrahydrocannabinol as related to genetic polymorphisms in CYP2C9. Clin Pharmacol Ther. 2009;85(3):273–6.
Bierut LJ et al. A genome-wide association study of alcohol dependence. Proc Natl Acad Sci U S A. 2010;107(11):5082–7.
Edenberg HJ et al. Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcohol Clin Exp Res. 2010;34(5):840–52.
Heath AC et al. A quantitative-trait genome-wide association study of alcoholism risk in the community: findings and implications. Biol Psychiatry. 2011;70(6):513–8.
Zuo L et al. Genome-wide association study of alcohol dependence implicates KIAA0040 on chromosome 1q. Neuropsychopharmacology. 2012;37(2):557–66.
Kendler KS et al. Genomewide association analysis of symptoms of alcohol dependence in the molecular genetics of schizophrenia (MGS2) control sample. Alcohol Clin Exp Res. 2011;35(5):963–75.
McGue M et al. A genome-wide association study of behavioral disinhibition. Behav Genet. 2013;43(5):363–73.
Treutlein J et al. Genome-wide association study of alcohol dependence. Arch Gen Psychiatry. 2009;66(7):773–84.
Frank J et al. Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict Biol. 2012;17(1):171–80.
Schumann G et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc Natl Acad Sci U S A. 2011;108(17):7119–24.
Gelernter J et al. Genome-wide association study of cocaine dependence and related traits: FAM53B identified as a risk gene. Mol Psychiatry. 2013;18:1211–7.
Gelernter J, et al. Genome-wide association study of opioid dependence: multiple associations mapped to calcium and potassium pathways. Biol Psychiatry. 2013.
Verweij KJ et al. The genetic aetiology of cannabis use initiation: a meta-analysis of genome-wide association studies and a SNP-based heritability estimation. Addict Biol. 2013;18(5):846–50.
Agrawal A et al. A genome-wide association study of DSM-IV cannabis dependence. Addict Biol. 2011;16(3):514–8.
Lubke GH et al. Estimating the genetic variance of major depressive disorder due to all single nucleotide polymorphisms. Biol Psychiatry. 2012;72(8):707–9.
Kos MZ et al. Common biological networks underlie genetic risk for alcoholism in African- and European-American populations. Genes Brain Behav. 2013;12(5):532–42.
Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacol. 2010;35(1):217–38. Reviews the role of the corticostriatal system in the onset and maintenance of addictions.
Bogdan R, Hyde LW, Hariri AR. A neurogenetics approach to understanding individual differences in brain, behavior, and risk for psychopathology. Mol Psychiatry. 2013;18(3):288–99. Reviews the emerging approach of combining genotype, neural correlates and behavior to study addictions and other psychopathology.
McClernon FJ et al. DRD4 VNTR polymorphism is associated with transient fMRI-BOLD responses to smoking cues. Psychopharmacol (Berl). 2007;194(4):433–41.
Franklin TR et al. Dopamine transporter genotype modulation of neural responses to smoking cues: confirmation in a new cohort. Addict Biol. 2011;16(2):308–22.
Kareken DA et al. A polymorphism in GABRA2 is associated with the medial frontal response to alcohol cues in an fMRI study. Alcohol Clin Exp Res. 2010;34(12):2169–78.
Villafuerte S et al. Impulsiveness and insula activation during reward anticipation are associated with genetic variants in GABRA2 in a family sample enriched for alcoholism. Mol Psychiatry. 2012;17(5):511–9. Links SNPs in GABRA2 which have been previously implicated in alcohol addiction to impulsivity and activation in the insula.
Janes AC et al. Association between CHRNA5 genetic variation at rs16969968 and brain reactivity to smoking images in nicotine dependent women. Drug Alcohol Depend. 2012;120(1–3):7–13.
Scult M, et al. Neural correlates of a genetic risk score for addiction, in Society for Neuroscience 2013: San Diego.
Hong LE et al. Association of nicotine addiction and nicotine's actions with separate cingulate cortex functional circuits. Arch Gen Psychiatry. 2009;66(4):431–41.
Hong LE et al. A genetically modulated, intrinsic cingulate circuit supports human nicotine addiction. Proc Natl Acad Sci U S A. 2010;107(30):13509–14.
Haughey HM et al. Marijuana withdrawal and craving: influence of the cannabinoid receptor 1 (CNR1) and fatty acid amide hydrolase (FAAH) genes. Addiction. 2008;103(10):1678–86.
Filbey FM et al. Marijuana craving in the brain. Proc Natl Acad Sci U S A. 2009;106(31):13016–21.
Filbey FM et al. Individual and additive effects of the CNR1 and FAAH genes on brain response to marijuana cues. Neuropsychopharmacol. 2010;35(4):967–75. Shows that variation in CNR1 and FAAH independently and additively are correlated with striatal activation during response to marijuana-related cues.
Schacht JP, Hutchison KE, Filbey FM. Associations between cannabinoid receptor-1 (CNR1) variation and hippocampus and amygdala volumes in heavy cannabis users. Neuropsychopharmacol. 2012;37(11):2368–76.
Solowij N et al. Cognitive functioning of long-term heavy cannabis users seeking treatment. JAMA. 2002;287(9):1123–31.
Tang DW et al. Genetic variation in CYP2A6 predicts neural reactivity to smoking cues as measured using fMRI. Neuroimage. 2012;60(4):2136–43.
Merikangas KR, Risch N. Genomic priorities and public health. Science. 2003;302(5645):599–601.
Thorsell A. The mu-opioid receptor and treatment response to naltrexone. Alcohol Alcoholism. 2013;48(4):402–8.
Ray LA et al. Subjective response to alcohol among alcohol-dependent individuals: effects of the mu-opioid receptor (OPRM1) gene and alcoholism severity. Alcohol Clin Exp Res. 2013;37 Suppl 1:E116–24.
Ramchandani VA et al. A genetic determinant of the striatal dopamine response to alcohol in men. Mol Psychiatry. 2011;16(8):809–17.
Anton RF et al. An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE) study. Arch Gen Psychiatry. 2008;65(2):135–44.
Maisel NC et al. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108(2):275–93.
Chen AC et al. Variation in Mu-Opioid Receptor Gene (OPRM1) as a moderator of naltrexone treatment to reduce heavy drinking in a high functioning cohort. J Alcohol Drug Depend. 2013;1(1):101.
Bauer LO et al. Variation in GABRA2 predicts drinking behavior in project MATCH subjects. Alcohol Clin Exp Res. 2007;31(11):1780–7.
Bauer LO, Covault J, Gelernter J. GABRA2 and KIBRA genotypes predict early relapse to substance use. Drug Alcohol Depend. 2012;123(1–3):154–9.
Johnson BA et al. Pharmacogenetic approach at the serotonin transporter gene as a method of reducing the severity of alcohol drinking. Am J Psychiatry. 2011;168(3):265–75.
Seneviratne C, Johnson BA. Serotonin transporter genomic biomarker for quantitative assessment of ondansetron treatment response in alcoholics. Front Psychiatry. 2012;3:23.
Kranzler HR et al. Sertraline treatment of co-occurring alcohol dependence and major depression. J Clin Psychopharmacol. 2006;26(1):13–20.
Kranzler HR et al. 5-HTTLPR genotype and daily negative mood moderate the effects of sertraline on drinking intensity. Addict Biol. 2013;18(6):1024–31.
Malaiyandi V et al. Impact of CYP2A6 genotype on pretreatment smoking behaviour and nicotine levels from and usage of nicotine replacement therapy. Mol Psychiatry. 2006;11(4):400–9.
Dempsey DA, et al. Genetic And Pharmacokinetic Determinants Of Response To Transdermal Nicotine In White, Black, And Asian Nonsmokers. Clin Pharmacol Ther. 2013;94(6):687–94.
Bergen AW et al. Nicotinic acetylcholine receptor variation and response to smoking cessation therapies. Pharmacogenet Genomics. 2013;23(2):94–103.
King DP et al. Smoking cessation pharmacogenetics: analysis of varenicline and bupropion in placebo-controlled clinical trials. Neuropsychopharmacology. 2012;37(3):641–50.
Nutt D et al. Development of a rational scale to assess the harm of drugs of potential misuse. Lancet. 2007;369(9566):1047–53.
Kreek MJ et al. Pharmacogenetics and human molecular genetics of opiate and cocaine addictions and their treatments. Pharmacol Rev. 2005;57(1):1–26.
Crist RC et al. An intronic variant in OPRD1 predicts treatment outcome for opioid dependence in african-americans. Neuropsychopharmacology. 2013;38(10):2003–10.
Fonseca F, et al. ALDH5A1 variability in opioid dependent patients could influence response to methadone treatment. Eur Neuropsychopharmacol. 2013.
Dickens C, Wiggins EO. A Dickens's tale of two cities. Boston: D. C. Heath and company; 1930. p. 392.
Barkley-Levenson AM, Crabbe JC. Bridging animal and human models: translating from (and to) animal genetics. Alcohol Res. 2012;34(3):325–35.
Kendler KS et al. Evidence for multiple genetic factors underlying the DSM-IV criteria for alcohol dependence. Mol Psychiatry. 2012;17(12):1306–15. Identified 3 independent genetic factors underpinning symptoms of alcohol dependence.
Lessov CN et al. Defining nicotine dependence for genetic research: evidence from Australian twins. Psychol Med. 2004;34(5):865–79.
Cubells JF et al. A haplotype at the DBH locus, associated with low plasma dopamine beta-hydroxylase activity, also associates with cocaine-induced paranoia. Mol Psychiatry. 2000;5(1):56–63.
Kosten T, et al. Vaccines against stimulants: cocaine and methamphetamine. Br J Clin Pharmacol. 2013.
Kosten TR et al. DBH gene as predictor of response in a cocaine vaccine clinical trial. Neurosci Lett. 2013;541:29–33.
Sharma L, Markon KE and Clark LA. Toward a theory of distinct types of "impulsive" behaviors: A meta-analysis of self-report and behavioral measures. Psychol Bull. 2013.
Volkow ND et al. The addictive dimensionality of obesity. Biol Psychiatry. 2013;73(9):811–8.
Wang S, et al. Introduction to deep sequencing and its application to drug addiction research with a focus on rare variants. Mol Neurobiol. 2013.
Vrieze SI, et al. Rare nonsynonymous exonic variants in addiction and behavioral disinhibition. Biol Psychiatry, 2013.
Xie P, et al. Deep resequencing of 17 glutamate system genes identifies rare variants in DISC1 and GRIN2B affecting risk of opioid dependence. Addict Biol. 2013
Duncan LE, Keller MC. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry. 2011;168(10):1041–9.
Keller MC, Gene x environment interaction studies have not properly controlled for potential confounders: The problem and the (simple) solution. Biol Psychiatry, 2013.
Higuchi S et al. Aldehyde dehydrogenase genotypes in Japanese alcoholics. Lancet. 1994;343(8899):741–2.
Meyers JL, et al. Childhood adversity moderates the effect of ADH1B on risk for alcohol-related phenotypes in Jewish Israeli drinkers. Addict Biol. 2013.
Johnson EO et al. Peer smoking and the nicotinic receptor genes: an examination of genetic and environmental risks for nicotine dependence. Addiction. 2010;105(11):2014–22.
Chen LS et al. Interplay of Genetic Risk Factors and Parent Monitoring in Risk for Nicotine Dependence. Addiction. 2009;104(10):1731–40.
Xie P et al. Childhood adversity increases risk for nicotine dependence and interacts with alpha5 nicotinic acetylcholine receptor genotype specifically in males. Neuropsychopharmacology. 2012;37(3):669–76.
Hartz SM et al. Increased genetic vulnerability to smoking at CHRNA5 in early-onset smokers. Arch Gen Psychiatry. 2012;69(8):854–60.
Nestler EJ. Epigenetic mechanisms of drug addiction. Neuropharmacology. 2014;76:259–68.
Zhang H et al. Profiling of childhood adversity-associated DNA methylation changes in alcoholic patients and healthy controls. PLoS One. 2013;8(6):e65648.
Bonsch D et al. Lowered DNA methyltransferase (DNMT-3b) mRNA expression is associated with genomic DNA hypermethylation in patients with chronic alcoholism. J Neural Transm. 2006;113(9):1299–304.
Zhang H et al. Hypermethylation of OPRM1 promoter region in European Americans with alcohol dependence. J Hum Genet. 2012;57(10):670–5.
Hillemacher T et al. Promoter specific methylation of the dopamine transporter gene is altered in alcohol dependence and associated with craving. J Psychiatr Res. 2009;43(4):388–92.
Xin Y et al. Genome-wide divergence of DNA methylation marks in cerebral and cerebellar cortices. PLoS One. 2010;5(6):e11357.
Taqi MM et al. Prodynorphin CpG-SNPs associated with alcohol dependence: elevated methylation in the brain of human alcoholics. Addict Biol. 2011;16(3):499–509.
Lessov-Schlaggar CN et al. Functional neuroimaging study in identical twin pairs discordant for regular cigarette smoking. Addict Biol. 2013;18(1):98–108.
Ripke S et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45(10):1150–9.
Crabbe JC, Bell RL, Ehlers CL. Human and laboratory rodent low response to alcohol: is better consilience possible? Addict Biol. 2010;15(2):125–44.
Devineni AV, Heberlein U. The evolution of Drosophila melanogaster as a model for alcohol research. Annu Rev Neurosci. 2013;36:121–38.
Lieberman R et al. Pilot study of iPS-derived neural cells to examine biologic effects of alcohol on human neurons in vitro. Alcohol Clin Exp Res. 2012;36(10):1678–87.
Acknowledgements
CHD receives support from T32DA007313. AA receives support from K02DA32573, R01DA23668 and R21AA021235.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Arpana Agrawal received grant funding, honoraria and travel reimbursements from ABMRF and has consulted for RTI International. She has received honoraria from NIDA and NIAAA. Arpana Agrawal’s spouse, Ryan Bogdan, is a co-author on this paper. Ryan Bogdan’s spouse, Arpana Agrawal, is a co-author on this study.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Demers, C.H., Bogdan, R. & Agrawal, A. The Genetics, Neurogenetics and Pharmacogenetics of Addiction. Curr Behav Neurosci Rep 1, 33–44 (2014). https://doi.org/10.1007/s40473-013-0004-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40473-013-0004-8