
Abstract
Background
Metagenomic analysis targeting the 16S rRNA gene has made it possible to characterize the vast array of microorganisms contained in the gut.
Aim
The purpose of this study was to evaluate how gut microbiota change in intensive care unit (ICU) patients in the acute phase after admission.
Methods
This prospective observational cohort study investigated 12 patients admitted to a single ICU of a large urban tertiary referral hospital. All patients were mechanically ventilated on admission. Fecal samples were collected from patients on days 1–2, 2–4, 5–8, and 7–10 after admission. DNA was extracted from fecal samples, and 16S rRNA deep sequencing was performed to monitor gut changes.
Results
Bacteria belonging to the phyla Firmicutes or Bacteroidetes were predominant in each sample. We observed serial dynamic changes in the percentages of Bacteroidetes and Firmicutes that were significantly altered during study period (p < 0.05). A ratio of Bacteroidetes to Firmicutes (B/F ratio) of >10 was seen in four of the six patients who died, whereas a B/F ratio of <0.10 was seen in only one of the six deaths. None of the survivors had a B/F ratio of >10 or <0.10. There was a statistical difference in the B/F ratio between the dead patients and survivors (p = 0.022).
Conclusions
Dynamic changes in gut microbiota at the phylum level of ICU patients during the acute phase were identified by high-throughput DNA sequencing. An extreme imbalance in gut microbiota may be associated with prognosis in critically ill patients.
Similar content being viewed by others


Introduction
The human gastrointestinal tract contains a diverse variety of microorganisms. Normal gut microbiota contain an estimated 1014 microbes [1] representing over 1000 different species of bacteria belonging to 190 different genera [2], the majority of which reside in the host colon [3]. They have important roles in human health, including metabolism and homeostasis of the immune system. Recently, many reports have revealed that changes in gut microbiota are related to metabolic [4], autoimmune [5], and chronic liver diseases [6] such as diabetes mellitus, inflammatory bowel diseases, and nonalcoholic steatohepatitis, respectively. Thus, disorder of the intestinal bacterial microbiota is connected with various diseases and is called “dysbiosis” [7]. Dysbiosis has been confirmed to occur not only in chronic diseases but also in acute illnesses. In the ICU setting, Shimizu et al. quantitatively evaluated the changes in gut microbiota and the environment in patients with systemic inflammatory response syndrome (SIRS). The patients with SIRS had 100–10,000 times fewer total anaerobes, including those of Bifidobacterium and Lactobacillus, and 100 times more Staphylococcus bacteria compared with those in healthy volunteers. The most dominant factors associated with mortality and septic complications were the numbers of total obligate anaerobes [8]. These findings indicate that more detailed investigation of obligate anaerobes, the dominant bacteria in commensal microbiota, should be performed to elucidate the role of microbiota in critically ill patients.
However, most microbes in the human body cannot be identified by culture methods because they are anaerobic and lose their symbiotic relationship once they are taken outside the body [9]. Thus, it is unclear how the composition of the whole gut microbiota changes during acute illnesses using conventional culture methods [10]. Metagenomic analysis targeting the 16S rRNA gene has been developed as a method to determine the microorganisms present in samples. This method is based on amplification of bacterial 16S rRNA genes and their analysis with massively parallel processing [11]. It has allowed researchers the opportunity to perform sequence-based studies of organisms and environments previously thought to be inaccessible, including obligate anaerobes and other microorganisms that cannot alive outside their hosts without symbionts [12].
The purpose of this study was to evaluate by a metagenomic procedure how gut microbiota change in intensive care unit (ICU) patients in the acute phase of critical illness.
Methods
Enrollment
This study was approved by the institutional review board of Osaka University Hospital. The study was conducted in the ICU of the emergency department of Osaka University Hospital, an academic urban tertiary referral hospital in Suita, Osaka, Japan, from February 2014 to August 2014. The ICU has 17 beds and treats 885 patients in 2014. Formal consent for participation in this study was obtained from each patient or their next of kin. Patients requiring mechanical ventilation at the time of admission were enrolled in this study. All patients were intubated. We included 12 patients in the study group. Exclusion criteria included postoperative patients with surgery of the rectum, patients with perianal infections, and patients expected to be on mechanical ventilation for <3 days. Seven healthy subjects were also included as a control group. They had never taken antibiotics and had not been admitted to hospitals within 6 months before participating in the current study.
Study Protocol
This study was an observational prospective cohort study. Enteral nutrition with Glucerna®-Ex (Abbott Japan Co., Ltd.) was started in the study subjects via nasogastric tube within 48 h of admission if there were no contraindications. An attending doctor determined both the class of antibiotics and duration of antibiotics use. Fecal samples were collected serially from patients during hospitalization on days 1–2, 2–4, 5–8, and 7–10 after admission. Fecal samples in the control group were collected serially in the same way. All samples were collected by inserting a sterile cotton-tipped swab 1–2 cm beyond the anus and rotating the swab for several seconds. Swabs were placed in sterile centrifuge tubes and immediately stored in a freezer at −78 °C until use. DNA was extracted from these fecal samples using a Power Soil DNA extraction kit (MOBIO). To monitor changes in gut microbiota in the study population, 16S rRNA deep sequencing was performed on an Ion PGM sequencer using a 318 chip and an Ion PGM Sequencing 400 Kit (both, Life Technologies). The resulting sequences were analyzed with the QIIME pipeline [13].
Patient Data Collection
Patient age, sex, Acute Physiology and Chronic Health Evaluation (APACHE II) score, and diagnosis were recorded on admission. The Sequential Organ Failure Assessment (SOFA) score was also recorded every day during hospitalization. Each patient’s final outcome and date of death were also archived.
Statistical Analysis
Sample size was computed based on feasibility. With 12 patients and 7 controls, the analysis was thought to have 85 % power at a two-sided significance level of 5 % to detect 1.5 standard deviation difference in the means of the outcome variables in which the standard deviation was for the corresponding outcome variable. Patient baseline demographic and clinical variables are presented as mean ± standard deviation. The composition of the five phyla of gut microbiota is presented as percentages. The absolute differences in percentage on each day from the percentage recorded on the first day of feces collection were used as variables to assess changes in gut microbiota. In order to account for dependencies in repeatedly measured observation within a subject, linear mixed-effect model was used to assess the differences between the patient and the control groups. Variance–covariance estimation was done with a basis on compound symmetry assumption. The mixed model was used in a similar manner way for the analysis of gut microbiota. Statistical analysis was performed using SPSS (version 22, SPSS, Chicago, IL). A significance level of two-sided 0.05 was used for statistical inferences.
Results
Patient characteristics are shown in Table 1. The study group comprised eight men and four women with a mean age of 66.9 ± 23.8 years. The admitting diagnosis was trauma in four patients, cardiac arrest in four patients, sepsis in three patients, and acute respiratory distress syndrome in one patient. APACHE II and SOFA scores at admission were 23.9 ± 12.4 and 7.1 ± 4.1, respectively. During the study period, each patient required mechanical ventilation and received systemic antibiotics intravenously. Enteral nutrition was administered within 48 h from admission in nine patients and was not administered in three patients due to complications with enteral feeding. Once enteral nutrition was started, it was continued until the patient was discharged or died. The course of antibiotics and the starting day of enteral nutrition are shown in Table 1. Six patients died due to exacerbation of the presenting disease, and the remaining six patients survived. The control group was comprised of seven men with a mean (±SD) age of 39.6 ± 8.3 years. There were two patients whose APACHE II scores were <10. In the case of patient 7, he suffered from cervical necrotizing fasciitis resulting from pharyngitis and required intubation to maintain his airway. His vital signs and laboratory data were almost normal on admission day. Patient 12 suffered from pelvic fracture, bladder injury, and urethral injury. At admission, although his circulation was not stable due to hemorrhage, other biochemical factors were almost normal. So, these patients’ APACHE II scores were not so high.
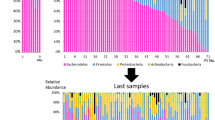
The taxonomic composition of the gut microbiota in each patient at the phylum level as determined by the metagenomic analysis of feces is shown in Fig. 1. The major five phyla of human gut microbiota are Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, and Fusobacteria. Bacteroidetes or Firmicutes was the predominant phylum in most patients at the first sampling on admission day, whereas Fusobacteria was predominant in only one patient. The composition of the gut microbiota was not stable in any of the patients, and dynamic changes were observed in all 12 patients.

Taxonomic composition of the gut microbiota at the phylum level as determined by metagenomic analysis. Color coding: blue = Bacteroidetes, red = Firmicutes, green = Proteobacteria, orange = Actinobacteria, yellow = Fusobacteria, gray = others. Hospital day of death is indicated by black circles on the time line
The relative proportion of each major phylum is shown in Fig. 2. The serial composition of Bacteroidetes decreased in three patients over the time course but increased in six patients. The absolute percentage difference from the first day of feces collection in the study group increased significantly compared with that in the control group (p = 0.014). The serial composition of Firmicutes decreased in six patients over the time course but increased in two patients. The absolute percentage difference from the first day of feces collection in the study group also increased significantly compared with that in the control group (p = 0.008). The serial compositions of Proteobacteria, Actinobacteria, and Fusobacteria in the study patients were not statistically different from those in the control group.

Serial changes of the major five phyla in the patient group and control group. The serial compositions of Bacteroidetes and Firmicutes changed significantly more in the critically ill patients than in the control group (p = 0.014, 0.018, respectively). The serial compositions of Proteobacteria, Actinobacteria, and Fusobacteria were not statistically different
Serial changes in the composition of gut microbiota were also evaluated by the ratio of Bacteroidetes to Firmicutes (B/F ratio). The changes occurring in the B/F ratio are shown in Fig. 3. Extremes of the B/F ratio were observed in five of the six patients who died. The B/F ratio was >10 in four of these six patients, whereas it was <0.10 in only one of the six. None of the survivors had a B/F ratio >10 or <0.10 during the study period. There was a statistical difference in the B/F ratio between the dead patients and the survivors (p = 0.022).

Serial changes in the ratio of Bacteroidetes to Firmicutes (B/F ratio) in the patient group and control group. A B/F ratio of >10 was seen in four of the six patients who died (black circles), whereas a B/F ratio of <0.10 was seen in only one of the six patient deaths. None of the survivors (triangles) had a B/F ratio >10 or >0.10 during the study period. There was a statistical difference in the B/F ratio between the dead patients and the survivors (p = 0.022)
Discussion
This is the first report, to our knowledge, to estimate the change in gut microbiota serially during the acute phase in ICU patients via 16S rRNA deep sequencing. The major findings of this study are that (1) the gut microbiota of ICU patients changes dynamically during the acute phase of severe illness, and (2) the changes in gut microbiota may be associated with patient prognosis.
The gut is an important target organ for stress after sepsis, trauma, burn, and shock [14]. Dysfunction of the intestinal epithelium, the immune system, and commensal bacteria in the gut is thought to cause the development of infectious complications and multiple organ dysfunction syndromes. The gut is the “motor” of multiple organ failure [15]. The use of antibiotics [16], vasoactive agents, agents to neutralize gastric secretions, sedatives or analgesic agents that impair intestinal motility [17], and diet [18] may correlate with changes in gut microbiota. In ICU settings, many patients are under severe insult from conditions such as trauma, sepsis, stroke, and cardiac failure, and they require many drugs. Their food intake and types of nutrition are also restricted. Thus, it is highly possible that disruptions of gut microbiota could occur in patients with critical illness during the acute phase. We previously reported alteration of the gut microbiota and environment in patients with severe SIRS [8]. However, the changes in gut microorganisms were evaluated by culture, which can detect and count only certain species of bacteria and cannot provide a complete picture of the gut microbiota. Metagenomic analysis has recently been developed that now makes it possible to see the overall picture. Zaborin et al. [19] reported by means of 16S rRNA analysis that the diversity of gut microbiota in humans significantly decreased, and pathogenic bacteria—the genera Enterococcus and Staphylococcus and the family Enterobacteriaceae—comprised the majority of gut microbiota during prolonged critical illness. The changes that occur in gut microbiota in the acute phase of critical illness, however, have yet to be thoroughly evaluated with metagenomics analysis. We believe that the present study is the first to assess the serial changes in gut microbiota in the acute phase of ICU patients by metagenomics analysis.
Bacteroidetes and Firmicutes, two major phyla of gut microbiota in humans, are involved in the regulation of lipid and bile acid metabolism to maintain energy homeostasis in the host [20]. Ley et al. [21] reported that the relative abundance of Bacteroidetes decreases and that of Firmicutes increases in obese people compared with lean people. Other reports also suggested similar changes in obese and lean patients [22]. However, another group reported opposite gut changes in obese patients, meaning that Bacteroidetes increased in obese patients [23]. Thus, there is some controversy regarding the role of Bacteroidetes and Firmicutes in gut microbiota. In addition, Mariat et al. [24] demonstrated that the ratio of Firmicutes to Bacteroidetes of the human microbiota changes with age. In the present study, the proportions of Bacteroidetes and Firmicutes changed significantly over the time course in the ICU, and extreme changes in the B/F ratio were observed in almost all of the patients with a poor prognosis. These results indicate that extreme changes in the composition of gut microbiota might influence or be associated with patient outcome. The reason for this alteration in gut microbiota was not clear, but there may be correlations with the antibiotics used, intestinal epithelial perfusion, and nutrition. It was also unclear whether the change in B/F ratio affected the metabolism or environment in the human intestine. Further study is needed to evaluate the factors that can disrupt gut bacterial microbiota and whether extreme alterations in gut microbiota are associated with patient prognosis.
This study has some limitations. First, it is a preliminary study and the sample size is too small to determine the cause of the changes in gut microbiota. Second, the metagenomic method used in this study can only evaluate the relative composition of microorganisms in samples, and thus we could not evaluate the absolute numbers of microorganisms in the patients in this study. Third, the analysis of gut microbiota was limited to the phylum level in this study. The main purpose of the current study was to evaluate the big-picture changes in gut microbiota. To understand changes in the gut more precisely, the gut microbiota must be analyzed at the subphylum level in a future study.
Conclusions
Dynamic changes in gut microbiota in ICU patients during the acute phase were identified by high-throughput DNA sequencing. An extreme imbalance of gut microbiota might be associated with prognosis in critically ill patients. The B/F ratio might be used in the future as a predictive factor of prognosis in the critically ill patient. Further study is needed to confirm these points.
References
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848.
Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65.
Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA. 1998;95:6578–6583.
Quigley EM. Gut bacteria in health and disease. Gastroenterol Hepatol (NY). 2013;9:560–569.
Kosiewicz MM, Zirnheld AL, Alard P. Gut microbiota, immunity, and disease: a complex relationship. Front Microbiol. 2011;2:180.
Compare D, Coccoli P, Rocco A, et al. Gut–liver axis: the impact of gut microbiota on nonalcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2012;22:471–476.
Festi D, Schiumerini R, Birtolo C, et al. Gut microbiota and its pathophysiology in disease paradigms. Dig Dis. 2011;29:518–524.
Shimizu K, Ogura H, Hamasaki T, et al. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig Dis Sci. 2011;56:1171–1177.
Feria-Gervasio D, Denis S, Alric M, Brugere JF. In vitro maintenance of a human proximal colon microbiota using the continuous fermentation system P-ECSIM. Appl Microbiol Biotechnol. 2011;91:1425–1433.
Shimizu K, Ogura H, Goto M, et al. Altered gut flora and environment in patients with severe SIRS. J Trauma. 2006;60:126–133.
Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180.
Tringe SG, Rubin EM. Metagenomics: DNA sequencing of environmental samples. Nat Rev Genet. 2005;6:805–814.
Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336.
MacFie J, O’Boyle C, Mitchell CJ, Buckley PM, Johnstone D, Sudworth P. Gut origin of sepsis: a prospective study investigating associations between bacterial translocation, gastric microflora, and septic morbidity. Gut. 1999;45:223–228.
Clark JA, Coopersmith CM. Intestinal crosstalk: a new paradigm for understanding the gut as the “motor” of critical illness. Shock. 2007;28:384–393.
Iapichino G, Callegari ML, Marzorati S, et al. Impact of antibiotics on the gut microbiota of critically ill patients. J Med Microbiol. 2008;57:1007–1014.
Rhee SH, Pothoulakis C, Mayer EA. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat Rev Gastroenterol Hepatol. 2009;6:306–314.
Schneider SM, Le Gall P, Girard-Pipau F, et al. Total artificial nutrition is associated with major changes in the fecal flora. Eur J Nutr. 2000;39:248–255.
Zaborin A, Smith D, Garfield K, et al. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. mBio. 2014;5:e01314–e01361.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023.
Bervoets L, Van Hoorenbeeck K, Kortleven I, et al. Differences in gut microbiota composition between obese and lean children: a cross-sectional study. Gut Pathog. 2013;5:10.
Collado MC, Isolauri E, Laitinen K, Salminen S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr. 2008;88:894–899.
Mariat D, Firmesse O, Levenez F, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123.
Funding
This work was supported by a Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Exploratory Research (Grant No. 26670788) and a medical research grant on traffic accidents from The General Insurance Association of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ojima, M., Motooka, D., Shimizu, K. et al. Metagenomic Analysis Reveals Dynamic Changes of Whole Gut Microbiota in the Acute Phase of Intensive Care Unit Patients. Dig Dis Sci 61, 1628–1634 (2016). https://doi.org/10.1007/s10620-015-4011-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-015-4011-3