
Abstract
Excessive body iron or iron overload occurs under conditions such as primary (hereditary) hemochromatosis and secondary iron overload (hemosiderosis), which are reaching epidemic levels worldwide. Primary hemochromatosis is the most common genetic disorder with an allele frequency greater than 10% in individuals of European ancestry, while hemosiderosis is less common but associated with a much higher morbidity and mortality. Iron overload leads to iron deposition in many tissues especially the liver, brain, heart and endocrine tissues. Elevated cardiac iron leads to diastolic dysfunction, arrhythmias and dilated cardiomyopathy, and is the primary determinant of survival in patients with secondary iron overload as well as a leading cause of morbidity and mortality in primary hemochromatosis patients. In addition, iron-induced cardiac injury plays a role in acute iron toxicosis (iron poisoning), myocardial ischemia–reperfusion injury, Friedreich ataxia and neurodegenerative diseases. Patients with iron overload also routinely suffer from a range of endocrinopathies, including diabetes mellitus and anterior pituitary dysfunction. Despite clear connections between elevated iron and clinical disease, iron transport remains poorly understood. While low-capacity divalent metal and transferrin-bound transporters are critical under normal physiological conditions, L-type Ca2+ channels (LTCC) are high-capacity pathways of ferrous iron (Fe2+) uptake into cardiomyocytes especially under iron overload conditions. Fe2+ uptake through L-type Ca2+ channels may also be crucial in other excitable cells such as pancreatic beta cells, anterior pituitary cells and neurons. Consequently, LTCC blockers represent a potential new therapy to reduce the toxic effects of excess iron.
Similar content being viewed by others

Introduction
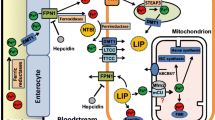
Iron is a transition metal that is essential for many biochemical, metabolic and biological processes. Iron is a component of hemoglobin, myoglobin, mitochondrial electron transport chain enzymes, cytochrome p450 system and many other proteins [1–3]. The biological importance of iron, as well as its toxicity, results from rapid oxidation–reduction cycling between ferric (Fe3+) and ferrous (Fe2+) states at physiological temperatures. Consequently, iron levels are precisely regulated under normal physiological conditions [1, 2] via several intricate feedback mechanisms involving transporters, iron-binding proteins and receptors (Fig. 1) [2]. Under several clinical conditions including primary hemochromatosis and secondary iron overload, iron metabolism is perturbed, which, combined with modifying environmental factors, leads to increased morbidity and mortality [4–7]. As a result of reductions in childhood mortality and increased use of blood transfusions, disease caused by iron overload is rapidly increasing in worldwide prevalence [4–7].

Cellular iron transporters and enzymes involved in iron uptake and export and the redox cycling of iron. DMT1 divalent metal transporter 1. Dcytb is a ferri-reductase, while ceruloplasmin and hephaestin are ferro-oxidases; broken arrow refers to the recycling of transferrin receptors
Under iron-overload conditions, iron in the circulation typically exceeds the capacity of iron binding by serum transferrin, leading to the appearance of highly reactive, non-transferrin-bound iron (NTBI) [8–10]. Uptake of NTBI into cells bypasses the normal negative-feedback mechanisms regulating cellular iron uptake and metabolism [2, 9–14]. Excess uptake of NTBI combined with the lack of an effective iron excretory pathway leads to the expansion of the labile intracellular iron pool (LIP) as well as the formation of highly reactive oxygen free radicals, causing peroxidation of membrane lipids and oxidative damage to cellular proteins [1, 2, 15]. Iron-mediated cellular damage plays a key pathophysiological role in multiple disorders including acute iron toxicosis [16, 17], iron-overload cardiomyopathy [18, 19], Friedreich-ataxia-associated cardiomyopathy [20], iron-overload endocrinopathies [21–24] and bone disease [25–27], myocardial ischemia–reperfusion injury [28, 29], atherosclerosis [30, 31] and neurodegenerative diseases [32, 33]. In this review, we will discuss the clinical disorders associated with iron overload, as well as the role of L-type Ca2+ channels (LTCC) in iron transport and pathophysiology of iron-overload cardiomyopathy and endocrinopathy.
Iron-overload conditions
Primary hemochromatosis
Primary hemochromatosis (hereditary or idiopathic) is a common inherited disorder that presents as four distinct subtypes (see Table 1) [1, 7]. In this condition, excessive iron accumulation results primarily from increased gastrointestinal absorption of iron that coupled with modifying environmental factors leads to iron overload [1, 2, 7]. Type 1 primary hemochromatosis (classic hereditary hemochromatosis) is an autosomal recessive disorder linked to mutations of the HFE gene that is involved in controlling gastrointestinal absorption of iron. Mutations of HFE genes involve either tyrosine for cysteine substitutions at position 282 (C282Y) [1, 7] or substitution of aspartate for histidine at position 63 (H63D) [1, 7]. The C282Y mutation is primarily limited to individuals of northern European ancestry and has an allele frequency of about 10%, while the H63D mutation occurs at allele frequencies greater than 5% in Mediterranean/Middle East regions and the Indian subcontinent [34]. There is a low and variable penetrance of the C282Y mutation with 0.5–1% of homozygotes developing frank clinical hemochromatosis [1, 7, 35]. Clinical effects of H63D mutations are generally limited to 1–2% of persons with compound heterozygosity for C282Y and H63D [7, 34].
Type 2 primary hemochromatosis is also called juvenile hemochromatosis and shows an autosomal recessive inheritance pattern. Patients with this disorder generally present at younger ages with larger iron burdens and severe organ damage before age 30 years (Table 1) [7, 36]. Most patients with juvenile hemochromatosis have mutations in the HJV gene (formerly called HFE2) that encodes for hemojuvelin, a protein with a central role in iron metabolism (see below) [7, 36]. A rarer form of juvenile hemochromatosis is caused by inactivation of hepcidin, another key regulator of iron homeostasis whose expression is modulated by hemojuvelin (Table 1) [7]. Type 3 primary hemochromatosis is another autosomal recessive condition that is linked to mutations in the transferrin receptor, TfR2 [7, 37]. In contrast to the other types of hemochromatosis, type 4 primary hemochromatosis is inherited in an autosomal-dominant manner and is linked to altered function of the iron exporter ferroportin, as discussed below (Table 1 and Fig. 1) [7, 38, 39]. Next to mutations in HFE, mutations in ferroportin are the most common cause of hemochromatosis [7, 38, 39].
In addition to inherited hemochromatosis, high dietary iron intake is also implicated in iron overload of sub-Saharan Africans, although a genetic component for this disease has also been implicated [40]. The presence of high serum ferritin (<200 μg/l) and normal transferrin saturation [41] is associated with NTBI and possible excess myocardial iron accumulation in some men without known genetic defects in iron metabolism.
Secondary iron overload
Unlike primary hemochromatosis that generally involves mutations in proteins involved in iron transport and metabolism, secondary iron overload occurs in patients with hereditary anemias including α-thalassemia [42], β-thalassemia [5] and sickle cell anemia [43]. In these patients, excessive iron exposure and secondary iron overload ensue primarily because of repeated blood transfusions coupled with increased gastrointestinal iron absorption in the setting of ineffective erythropoiesis [1, 2, 5, 44]. A reduction in childhood mortality (due to infection and malnutrition) combined with increased use of chronic blood transfusions has led to a growing incidence of iron overload in patients with hereditary anemia [4, 6, 43, 45]. α- and β-thalassemias are caused by mutations resulting in defective synthesis of the α- and β-globin chains of hemoglobin, respectively, and are the most common monogenetic diseases in humans (Table 1) [5, 6, 42]. Patients with thalassemia originate primarily from the Mediterranean regions, the Indian subcontinent and Southeast Asia, where the estimated gene frequency is typically 3–10% but can be as high as 30–40% within certain subpopulations [5, 6, 42]. The clinical manifestations of thalassemia range from the silent carriers to severe iron overload. Sickle cell anemia is the most common and severe form of sickle cell disease (SCD) caused by homozygous presence of a glutamate to valine mutation in the beta-globulin gene (Hgb S) (Table 1). Sickle cell anemia occurs most commonly in individuals of African ancestry, and in the United States, 9% of African Americans carry the sickle cell trait and one in 600 has sickle cell anemia [46, 47].
In addition to thalassemia and SCD, several other clinical disorders are associated with secondary iron overload including sideroblastic anemia, myelodysplastic syndrome, acute myeloid leukemia, congenital dyserythropoietic anemia and chronic renal failure (Table 1). Sideroblastic anemia is characterized by mitochondrial iron overload in erythroblast and is associated with systemic iron overload, suggesting an important role for mitochondria in cellular iron metabolism (Table 1) [44, 48]. In patients with chronic renal failure and end-stage kidney disease, anemia is common due in part to erythropoietin deficiency that often necessitates therapy with intravenous iron [49–51]. A growing recognition of the toxicity of excess iron levels has increased concerns regarding the use of parenteral iron [49] in treating these patients [50, 51].
Acute iron poisoning
Acute iron toxicosis is a common cause of pediatric drug-overdose mortality [52–54]. Acute iron poisoning in these patients leads to gastrointestinal, liver and myocardial injury coupled with microvascular dysfunction, and shock [52–54]. Mortality observed with acute iron toxicity is linked to cardiovascular effects that include impaired myocardial contractility, bradycardia and hypotension, as discussed further below [16, 17].
Iron metabolism and transport
Transport of iron
The human body uses about 20 mg of iron per day for hemoglobin synthesis and the production of approximately 200 billion erythrocytes [1, 2]. Consequently, most of the iron in the body is contained in hemoglobin. An additional 4–5 mg of iron is utilized daily for the production of other cellular proteins such as mitochondrial proteins, muscle myoglobin and others. A significant amount of iron is also stored at relatively high levels in hepatocytes and macrophages of the reticulo-endothelial system. Hepatocytes extract dietary iron delivered from the intestines, while reticulo-endothelial macrophages recycle iron from ingested senescent red blood cells. Once in the body, iron is delivered to most cells of the body via the blood as ferric iron bound to the abundant serum protein transferrin (i.e. diferric-transferrin). Despite large daily iron fluxes within the body, the total iron content in normal individuals is relatively fixed, with men (50 mg Fe/kg) having slightly higher iron levels than women (40 mg/kg).
Movement of iron within the body is mediated by a combination of transporters whose activity is complexly regulated and modulated by various factors [2]. The best described “traditional” transporters include dimetal transporter 1 (DMT1), transferrin receptors (TfR1 and TfR2), ferroportin and the putative “heme receptor”. Our recent studies have established that iron can also be transported in excitable cells like cardiomyocytes by voltage-dependent L-type Ca2+ channels, which are promiscuous divalent transporters. Generally, iron transport involves redox cycling between ferrous (Fe2+) and ferric (Fe3+) states (Fig. 1 and Table 2) that are catalyzed by several ferric reductases such as duodenal-cytochrome b (Dcytb) as well as ferroxidases such as ceruloplasmin and hephaestin (Fig. 1) [2, 7]. Both ceruloplasmin and hephaestin are multi-copper-containing ferroxidases that are present in the serum and as a transmembrane protein in the basolateral surface of the duodenum, respectively, and are necessary for iron egress from intestinal enterocytes into the circulation and binding to transferrin [2, 55].
Transferrin–transferrrin receptor system
The best characterized and most prevalent method for iron transport involves the binding of plasma diferric-transferrin iron (i.e. transferrin-bound iron) to type I or type II transferrin receptors (i.e. TfR1 or TfR2). In individuals with normal iron levels, plasma iron is almost exclusively bound to transferrin (Tf), an abundant plasma protein with an extraordinarily high affinity for ferric iron at normal plasma pH [1, 2]. Transferrin-bound ferric iron is relatively “non-reactive” and is extracted from the plasma by most cells of the body following binding of ferritin to transferrin receptors (TfR1 and TfR2) in clathrin-coated regions of the cell membrane [1, 2]. Complexes of transferrin and TfR1/2 are internalized into acidic endosomes where the iron is reduced (to Fe2+) and released into the cytosol by the divalent metal transporter 1, DMT-1 (see below) [2]. While the type 1 transferrin receptor, TfR1, is ubiquitously expressed, the TfR2 expression is restricted to hepatocytes, duodenum and erythroid precursors [2, 56]. Both receptors are expressed at relatively high levels in hepatocytes of the liver and enterocytes of the small intestine, especially the duodenum. The expression of TfR1 is profoundly down-regulated under iron-overload conditions, particularly in the liver, through the action of iron-response elements [1, 2]. By contrast, TfR2 protein expression is up-regulated when iron is elevated, making TfR2 a potential sensor or marker of iron status [56, 57]. TfR2 up-regulation is probably an important contributor to excessive hepatic iron uptake in iron overload [56, 57].
Divalent metal transporter 1
In addition to transferrin-bound iron, iron also enters cells by DMT1 transporters (also called Nramp2, DCT1 or SLC11A2), which are H+/divalent metal symporters capable of transporting many other divalent metals including Mn2+, Cu2+, Zn2+ and Cd2+ [58–60]. DMT1 is highly expressed in the kidney and intestines while being expressed at much lower levels in brain and heart [58–60]. In the brush borders of the duodenum, DMT1 transports iron into enterocytes from the intestinal lumen following reduction in dietary non-Tf-bound ferric to ferrous iron by intestinal ferric reductase Dcytb, a process that is facilitated by low pH in the duodenum where most of the dietary iron is absorbed (Fig. 1) [58–60]. As already mentioned, DMT1 also mediates iron export from the intracellular endosomes following iron uptake via the transferrin and heme uptake systems.
Heme–iron transport
Iron can also be imported as a heme–iron complex into selected tissues such as intestinal enterocytes and reticulo-endothelial macrophages by a putative “heme receptor”, whose molecular identity remains unclear. A membrane receptor called feline leukemia virus type C receptor (FLVCR) can facilitate transport of heme into erythrocytes and may also impact on heme transport in the intestine and liver [61]. Once internalized, heme-bound iron is released by heme oxygenase.
Ferroportin
Ferroportin (Fpn), a divalent iron export protein that is also known as iron-regulated protein (IREG1), metal-transporter protein (MTP1) or SLC11A3, is highly expressed on the surface of enterocytes, macrophages, hepatocytes and placental cells [7, 38, 39]. In these cells, Fpn exports reduced iron into the plasma, whereupon the iron is oxidized by ceruloplasmin or hephaestin, which are copper-containing ferroxidases found in the serum as well as the basolateral surface of enterocytes. Once oxidized, iron binds to transferrin [2, 55]. Fpn levels are regulated by the binding of hepcidin, a critical iron regulatory protein (see below), which leads to Fpn internalization and degradation, thereby decreasing export of cellular iron [38, 39]. As already mentioned, type 4 primary hemochromatosis is associated with Fpn mutations leading to impaired export of cellular iron and the subsequent accumulation of iron and iron overload (see Fig. 1) [7, 38, 39]. There are multiple Fpn mutations leading either to defective cell surface localization, decreased ability to be internalized and degraded in response to hepcidin, or dominant negative mutants, which may explain the phenotypic variation as well as the dominant inheritance of the disease [38, 39]. In addition to controlling the exit and release of intracellular iron, ferroportin also controls the intestinal uptake of extracellular iron at the apical membrane, possibly by modulating the activity of DMT1 [62].
Non-transferrin bound iron
Although iron absorption is normally tightly regulated in order to maintain iron at levels that prevent tissue damage due to oxidative stress, elevated body iron does occur under several disease conditions due to excessive intestinal iron absorption or repeated blood transfusions leading to surplus iron accumulation in tissues such as liver, spleen, bone marrow, pancreas, heart, pituitary and central nervous system. As transferrin becomes iron-saturated, iron increasingly resides as NTBI, which is readily transported into selected tissues of the body, particularly heart, endocrine tissue and brain [8, 9]. In patients with secondary iron overload, total serum iron levels range from 20 to 60 μM (normal range=8–15 μM) with estimated NTBI levels ranging from 1 to 10 μM [2, 8, 9, 63]. Excessive iron levels in the cell saturate the ferritin binding capacity, leading to iron binding to low molecular weight compounds that form the LIP. This iron is highly catalytically active and participates in free-radical-generating reactions [2, 14].
Although the mechanism for the transport of NTBI into cells remains unclear, it is clear that selected tissues are particularly susceptible to excess iron uptake when NTBI is present. Several transport mechanisms have been proposed to participate in NTBI uptake. Recent evidence suggests that TfR2 is responsible for the excessive iron uptake into liver hepatocytes since TfR2 expression levels increase in iron overload, leading to liver cirrhosis and hepatic tumors [56, 57]. However, NTBI uptake into excitable cells such as cardiomyocytes, neurons, beta cells of the pancreas and pituitary cells requires reduction of ferric iron (Fe3+) to ferrous iron (Fe2+) by a membrane-associated ferri-reductase system, properties that clearly distinguish it from transferrin-receptor-mediated ferric iron uptake [11, 12, 64]. In these excitable tissues, it has been suggested that NTBI uptake is DMT1-dependent [59]. However, the maximum NTBI uptake rate (i.e. V max) in cardiomyocytes increases without changes in sensitivity to iron (i.e. K D), which contrasts with DMT1 whose protein expression decreases when iron is elevated [60]. In addition, DMT1 is expressed at extremely low levels in most excitable tissue including the heart [59, 60]. We have shown that NTBI uptake into cardiomyocytes is dependent on L-type Ca2+ channels, which are known to be promiscuous divalent metal transporters [65–68]. Importantly, L-type Ca2+ channels satisfy all the criteria consistent with the NTBI transporter, and most importantly, this transporter readily explains the pattern of tissues at risk for damage and dysfunction under iron-overload conditions. Specifically, tissues with the greatest risk in iron overload are excitable tissues with high duty cycles for L-type Ca2+ channel activity: cardiomyocytes, anterior pituitary cells, pancreatic beta cells and neurons (see Table 3). Evidence supporting a critical role for NTBI uptake by L-type Ca2+ channels in cardiomyocytes of the heart is reviewed below.
Mechanisms for the regulation of iron
Iron homeostasis necessitates tight control of iron uptake, storage and export as well as management of intracellular iron distribution [1, 2]. A major requirement for proper iron homeostasis is limiting the labile intracellular iron while simultaneously ensuring iron availability for many essential iron-containing proteins and enzymes. Since the loss of iron from the body is, under normal circumstances, largely independent of the total body iron, alterations in total body iron are primarily set by modulation of its absorption in the small intestines, especially in the duodenum. Iron uptake by the small intestine appears to be regulated by two interrelated processes: crypt programming and hepcidin expression levels [2, 7]. Crypt programming involves setting the iron uptake rates in mature villus enterocytes based on the total body iron levels sensed by developing enterocytes within the intestinal crypts via TfR1/2. Specifically, iron levels in the developing enterocytes influences the expression pattern of various iron transporters. Intestinal iron uptake also appears to be regulated in immature enterocytes by HFE, the gene involved in type I hemochromatosis [7]. With mutations in HFE in human (as well as in HFE knockout mice), transferrin-bound iron uptake is decreased in association with enhanced expression and activity of FPN1, resulting in increased iron export from enterocytes into the blood [69, 70]. Regulation of iron uptake in enterocytes may also involve beta 2-microglobulin that forms a macromolecular complex with HFE and TfR1.
Modulation of iron transport in small intestinal enterocytes and reticulo-endothelial macrophages by hepcidin represents an alternate mechanism for regulating iron. Hepcidin is a small (20–25 amino acids) cysteine-rich antimicrobial peptide that is expressed and secreted into the blood by the liver in response to elevated iron and inflammation [2, 7, 36]. Hepcidin inhibits iron excretion in macrophages and enterocytes by binding to Fpn1, leading to internalization and loss of function of Fpn1 [38]. By contrast, in iron deficiency and anemia as well as in type 1 primary hemochromatosis, hepcidin levels are reduced, which increases Fpn1 expression as well as iron release from enterocytes and macrophages [70]. Indeed, loss of HFE function in type 1 primary hemochromatosis impairs the ability of hepcidin to regulate duodenal iron absorption, which may be the mechanism responsible for excessive iron absorption under this condition [70, 71]. Thus, post-translational regulation of Fpn1 by hepcidin forms a homeostatic loop whereby iron regulates secretion of hepcidin, which in turn controls the concentration of Fpn1 and thereby iron export at the enterocyte surface [38, 72]. This loop also operates in the regulation of iron levels under inflammatory conditions, which may explain the anemia in chronic inflammation as well as the antimicrobial actions of hepcidin [2, 7, 70, 73]. Although the precise physiological role of hemojuvelin (HJV; also known as RGMc and HFE2), a glycosylphosphatidylinositol-anchored protein, remains to be elucidated, recent experimental and clinical studies indicate that HJV is involved in the inflammation-induced increases of hepcidin expression [7, 36, 74, 75]. This connection helps explain the central role of hepcidin (and HJV) in the pathogenesis of juvenile hemochromatosis as well as anemia in chronic diseases (see Table 1) [7, 36, 74, 75].
The regulation of iron crypt programming and hepcidin, as well as the regulation of iron uptake in many cells of the body, involves tight negative-feedback regulation [2, 7, 9, 58] via iron regulatory proteins (IRP1 and IRP2) that recognize iron-responsive elements (IRE) located on the 3′ or 5′ untranslated regions (UTR) of mRNA for many iron-regulated genes. For example, the transferrin receptors and H and L subunits of the intracellular iron-storage protein, ferritin [76, 77], contain IREs in the 3′-UTR and 5′-UTR of their mRNAs, respectively [2, 78]. Under conditions of low iron, IRP binding to IREs blocks translation of ferritin mRNA and stabilizes transferrin receptor mRNA. When labile intracellular iron increases, IRP1 binding to IREs leads to IRP2 degradation, resulting in efficient translation of ferritin and rapid degradation of transferrin receptor mRNA [2, 77, 78]. DMT1 and FPN1 also contain IREs in their mRNA UTRs that are suggested to be responsible for the decreased DMT1 expression in the heart (and intestine) observed with iron overload (Table 2) [2, 7, 59, 60].
Iron-overload cardiomyopathy
Clinical description
Iron-overload cardiomyopathy is a common cause of death due to cardiovascular complications especially in the second and third decades of life [4–6, 79, 80]. Indeed, in European [81], North American [79, 80] and Chinese [24] patients with thalassemia major, the severity of cardiac dysfunction is the primary determinant of survival, while in patients with primary hemochromatosis, cardiovascular disease also contributes significantly to their mortality and morbidity [82–85]. In addition, cardiac iron accumulation in beta-thalassemia patients correlates directly with both heart disease and mortality [5, 79, 80]. Although iron chelation therapy is widely used for treating iron-overload patients, cardiomyopathy and mortality are still common in these patients [24, 86]. Regardless of its origin, iron overload in patients [13, 87, 88] and animal models [18, 19] leads to a restrictive cardiomyopathy with prominent early diastolic dysfunction that invariably progresses to end-stage dilated cardiomyopathy characterized by impaired systolic function and reduced mean arterial blood pressure often accompanied by arrhythmias including atrioventricular (AV) block, conduction defects, bradyarrhythmias, tachyarrhythmias and sudden cardiac death (Fig. 3) [79, 80, 84, 89–92]. AV nodal block is a particularly common occurrence, which is due to an especially high accumulation of iron in the AV nodal compared to other tissues of the heart (see Fig. 3 and Table 3) [89].
Pathophysiology
The precise mechanism underlying cardiomyocyte dysfunction induced by iron overload is not entirely clear. Iron within cells can be divided into the “unavailable” or tightly bound pool, which includes iron in ferric hydroxide precipitates and iron complexed with ferritin, and the “available” LIP. The LIP is more readily available for Fenton-type reactions whereby the conversion of reduced iron (Fe2+) into oxidized iron (Fe3+) generates various free radicals [15, 93]. Under normal physiological conditions, the generation of free radicals is minimized by a repertoire of enzymatic and non-enzymatic antioxidant mechanisms [93]. However, when iron levels are chronically elevated, excessive free radical generation leads to depletion of antioxidants and increased cellular damage due to oxidation of lipids, proteins and nucleic acids [19, 93]. in animal models [18, 19, 94] as well as in patients with primary hemochromatosis, beta-thalassemia major and end-stage kidney disease [95–97]. Oxidation of lipids (i.e. lipid peroxidation) is of particular importance in iron overload that leads to marked increases in unsaturated (malondialdehyde and hydroxynonenal) and saturated (hexanal) aldehydes in the heart and plasma [18, 19]. These compounds are associated with cellular dysfunction, cytotoxicity and cell death [18, 19, 93, 98, 99]. Consistent with a critical role of oxidative stress in iron-overload cardiomyopathy, the risk of developing iron-overload cardiomyopathy in patients with primary hemochromatosis is strongly influenced by polymorphisms of the manganese-superoxide dismutase (MnSOD) gene [100]. Additional factors that may also modulate the amount of oxidative stress observed under iron-overload conditions include inflammation [101, 102] and ApoE polymorphisms (indicator of low intrinsic antioxidant status) [103].
Iron-induced oxidative damage can have multiple effects on cardiac function. For example, iron overload has been linked to cardiomyocyte loss due to apoptosis [19, 29, 93], which can be reduced by antioxidant treatment [19]. Enhanced apoptosis and/or necrosis in iron-overload cardiomyopathy could be linked to altered mitochondrial function as seen in Friedreich ataxia [93, 104, 105]. Increased myocyte loss is probably a major contributor to the prominent interstitial fibrosis seen in iron-overload cardiomyopathy, although iron-mediated effects on cardiac fibroblasts might also contribute [9]. Regardless, reduced cardiomyocyte numbers combined with increased myocardial interstitial fibrosis are predicted to contribute to impaired systolic and diastolic function in iron-overloaded hearts. Increased iron-mediated oxidative stress can also directly alter myocyte electrical and contractile properties by affecting the function of several regulators of cardiac excitation–contraction coupling [106–108]. Specifically, with elevated oxidative stress, sarcoplasmic reticulum (SR) Ca2+ leak through ryanodine receptors (Ca2+ release channels) [109] is increased, sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) activity is inhibited, sodium–calcium exchanger (NCX) [110] currents are elevated, LTCCs are reduced [111, 112] and sodium as well as potassium currents can be either enhanced or depressed (see Fig. 2) [113]. These effects can occur by direct protein oxidation or by the generation of lipid peroxidation products [114, 115]. Collectively, elevated oxidative stress leads to reduced peak systolic Ca2+ levels, slowed rates of Ca2+ relaxation and elevated diastolic Ca2+ levels, which probably contribute to the impaired systolic and diastolic function observed in iron-overload cardiomyopathy and also leads to arrhythmias, impaired electrical conduction and sudden death [116].

Interaction between iron-mediated oxidative stress and the excitation–contraction coupling in a cardiomyocyte. ROS reactive oxygen species, SERCA2a sarcoplasmic reticulum Ca2+ATPase isoform 2, NCX sodium–calcium exchanger, SR sarcoplasmic reticulum
Altered function of ion channels and electrogenic transporters in response to iron overload and elevated oxidative stress can lead to increased susceptibility and generate arrhythmias in the heart [113, 116, 117], which are commonly observed under iron-overload conditions [79, 113, 116]. For example, spontaneous Ca2+ release combined with slowed Ca2+ re-uptake in the SR and increased NCX activity predisposes to increased triggered arrhythmias and delayed after-depolarization that are observed under iron-overload conditions. In iron-overloaded gerbils, ventricular action potential duration has been shown to be abbreviated in connection with increased transient outward K+ current (I to) densities coupled with reduced Na+ currents [113], although reduced L-type Ca2+ currents might also contribute. In iron-treated mice and gerbils, common electrophysiological observations include QRS prolongation accompanied by slowed conduction velocity, bradycardia and prolongation of the PR interval in association with first- and second-degree AV block (Fig. 3) [92]. Slowed conduction in the heart may be related to the observation that elevated oxidative stress reduces cardiac voltage-gated Na+ currents [118]. Action potential shortening and abnormal impulse conduction [92, 113] in the setting of increased interstitial fibrosis observed in iron overload [18, 19] increase the propensity for unidirectional conduction block, wavefront breakup and creation of arrhythmic ventricular re-entry circuits [92, 117]. The reduced heart rates and AV block may be related to a combination of preferential iron deposition and increased interstitial fibrosis in these regions [18, 89–91]. In addition, iron deposition in the myocardium is heterogeneous, which can further promote arrhythmias and sudden cardiac death by increasing the heterogeneity of ventricular repolarization and QT dispersion, which is linearly correlated with iron burden [89, 113, 119].

Telemetric recordings from conscious mice that were injected with placebo or iron (n=6) over a 4-week period as previously reported [18]; baseline heart rate=500–600 bpm. a Normal ECG tracing. b Iron-overload-induced first-degree AV block (PR interval=42±2.1 vs 89.2± ms; p<0.01) and sinus arrest (sick SA node). c Iron-overload-induced AV block as illustrated by Mobitz type II block and conduction delay with widened QRS complex (QRS duration=16.4±0.6 vs 41.2±4.9 ms; p<0.01). d Progressive bradycardia in an iron-overloaded mouse that occurred over a 5-day interval culminated into an idioventricular rhythm and death; mv millivolt, bpm beats per minute
In addition to the direct effects on the heart, elevated iron can also affect the function of vascular smooth muscle cells and endothelial cells. Indeed, endothelial dysfunction and increased arterial stiffness are commonly observed in patients with primary hemochromatosis [120], beta-thalassemia major [121] and sickle cell anemia [122]. These vascular changes can in turn affect myocardial perfusion and function.
NTBI transport by L-type Ca2+ channels in heart
The basis for the high sensitivity of the heart (and other excitable tissues) to excessive iron uptake, damage and dysfunction under conditions of iron overload was an enigma for many years. However, several classical characteristics of NTBI transport and of the cardiomyopathy under iron-overload conditions provide important insights into the possible mechanism of iron transport into the heart under these conditions. For example, NTBI transport in cardiomyocytes requires divalent ferrous, not trivalent ferric, iron, and its rate increases markedly with chronic iron elevations [12, 64, 68]. These two features argue strongly against TfR-mediated or DMT1-mediated mechanisms for NTBI transport in the heart. Specifically, TfR1 transports ferric iron and TfR1 mRNA expression is reduced in association with a 63% decline in IRP1 in cardiomyocytes exposed to chronic elevations in iron [123]. Similarly, while DMT1 can transport ferrous iron, myocardial DMT1 mRNA expression has been shown either to decrease with elevated iron in adult hearts [60] or to be uncorrelated with changes in iron accumulation in cultured myocytes despite the absence of mRNA changes [123]. The lack of involvement of these iron transporters in myocardial NTBI uptake is further supported by low-level TfR1-dependent iron uptake into heart as well as the extremely low expression levels of TfR1 and DMT1 in the heart [60, 123].
Unlike DMT1 and TfR, L-type Ca2+ channels satisfy all the known properties of NTBI transport. Indeed, while LTCCs are primarily utilized for the transport of Ca2+, they are able to transport many other divalent, but not trivalent, cations including Fe2+, Zn2+, Co2+, Sr2+, Ba2+ and Mn2+ [66–68, 124]. Consistent with a dominant role of LTCC in myocardial iron transport, iron uptake is increased by the selective LTCC agonist, Bay K 8644, as well as following beta-adrenergic stimulation in proportion to the degree of LTCC current enhancement while being inhibited by LTCC blockers [68]. Moreover, low Fe2+ levels slow the rate of LTCC current inactivation, resulting in increased Fe2+ entry per heartbeat [68], and L-type Ca2+ currents are not decreased in iron overload [18], consistent with the absence of IREs in the mRNA of the pore-forming subunits of cardiac LTCCs (i.e. CaV1.2) (see Table 2). The Fe2+-mediated slowing of Ca2+ current inactivation could arise from competition between Fe2+ and Ca2+ for the C-terminal cytoplasmic Ca2+ binding site involved in Ca2+-mediated inactivation of LTCCs [108]. Thus, LTCCs are the only Fe2+ transporter whose activity increases with elevated iron, consistent with NTBI uptake in the heart under iron-overload conditions [9, 12, 18, 68]. Remarkably, the amount of myocardial iron accumulation predicted to occur via LTCCs over 20 years, a relevant period for patients with iron overload, is estimated to be about 10 mg of iron per gram of myocardial tissue [68], which matches closely to the 3–10 mg/gm of myocardial iron measured in patients with iron-overload cardiomyopathy [89] and in animal models [18, 19].
A more definitive link between LTCCs and myocardial iron deposition was obtained in mouse studies where LTCC blockers, like amlodipine and verapamil, reduced intracellular myocardial iron accumulation and reduced oxidative stress while protecting diastolic and systolic cardiac function [18]. Total myocardial iron level was determined by atomic absorption spectrophotometry, while intracellular iron deposition was examined by Prussian blue histological staining coupled with analytical electron microscopy [18, 19]. Moreover, cardiac-specific LTCC overexpression caused increases in myocardial iron accumulation and oxidative damage in proportion to the elevated Ca2+ currents that were, again, reduced with LTCC blockers [18]. These beneficial cardiac effects of LTCC blockers in iron-overload mice were not observed in the liver, as would be expected, since hepatocytes express minimal levels of functional LTCCs [125]. Involvement of LTCC in iron transport is also supported by the protective actions of taurine (i.e. 2-aminoethane-sulphonic acid) supplementation against myocardial iron accumulation, oxidative damage, and altered cardiac structure and function [19]. The link between LTCCs and iron uptake in excitable tissues is consistent with the observation that the maximal rates of iron uptake (V max) are approximately tenfold higher in cardiomyocytes than in fibroblasts [12, 64, 123] that have few if any LTCCs. The link between LTCC and myocardial iron transport is further substantiated by the high susceptibility to iron-mediated injury and disease observed in excitable cells that require high LTCC activity for proper functioning such as excitation–contraction coupling in vascular smooth muscle cells [106–108] as well as in excitation–secretion coupling in osteoblasts [126], endocrine cells and neurons [106, 127–129].
The molecular basis for iron permeation in LTCC has not previously been examined. However, ion selectivity, permeability and blocking properties of LTCCs are dictated by electrostatic binding of ions within the pore [130, 131]. Strong electrostatic interactions between ions and the selectivity filter combined with a large pore radius (i.e. 6.2 nm) allows many divalent cations to readily permeate LTCCs, independent of their ionic radius (i.e. r Ca=2.4 nm vs r Fe=2 nm), although some permeant ions also “block” LTCC currents depending on the relative binding affinity [65, 124]. By contrast, under normal biological conditions, monovalent cations cannot easily permeate LTCC, while trivalent cations are often potent blockers of LTCCs. These biophysical properties of cardiac LTCCs originate from the pore-forming α1 subunits that are comprised of four internal repeat domains (I–IV) each made up of six membrane-spanning segments (S1–S6) [106, 108, 132] and which co-assemble with β-subunits and α2/δ-subunits to form functional LTCCs [106, 108]. The cardiac α1 subunits are members of a highly homologous family of high-voltage-activated Ca2+ channel genes (i.e. the Cav1 family) that includes Cav1.1, Cav1.2 (α1C) and Cav1.3 (also called α1S, α1C and α1D) [106, 108, 132] Cardiomyocytes express Cav1.2 [128, 133, 134], which is also expressed (as spliced variants) in vascular smooth muscle [128], brain [128] and endocrine tissue including pancreatic beta cells [127, 128]. Cav1.3 shows a far more restricted expression pattern, being found in sinoatrial (SA) nodal cardiomyocytes [133] as well as pancreatic beta cells where it plays a relatively minor role in insulin release [128]. Cav1.1 subunits are expressed in skeletal muscle [106]. Given the high degree of amino acid identity and thereby structural similarity between the Cav1 family members, it seems likely that LTCCs also participate in NTBI uptake in excitable tissues other than the heart. Indeed, as already mentioned, the cells most susceptible to damage and dysfunction in iron overload are precisely those having high densities of LTCC. One potential glaring exception to this pattern is skeletal muscle cells, which have high densities of LTCC. However, skeletal muscle cells utilize LTCCs for excitation–contraction coupling without requiring ion permeation [106], consistent with the relative resistance of skeletal muscle to the effects of iron overload.
Involvement of LTCC in iron transport helps explain several key clinical features of iron-overload cardiomyopathy. For example, second- and third-degree heart blocks are commonly observed in iron overload, and these patients often require pacemaker device implantation [89–91]. Prominent AV node pathology is consistent with the essential requirement of LTCC in AV nodal conduction [135] that is reflected in the very high baseline LTCC current densities recorded in AV nodal myocytes (i.e. 14–18 pA/pF) [136, 137]. Iron transport by LTCC could explain the observation that the L-type Ca2+ channel blocker diazepam protects the heart against acute iron-induced toxicity and mortality without affecting iron absorption and excretion [138, 139]. Similarly, Ca2+ channel blockers protect the myocardium from ischemia–reperfusion injury [140, 141], which has been linked to iron-mediated injury [29, 142].
NTBI entering cardiomyocytes is effectively trapped in the cytosol following rapid redox cycling to ferric iron and binding to ferritin [76, 143], conversion to the insoluble ferric iron (hemosiderin) deposits [113] or expansion of the reactive LIP leading to iron-mediated oxidative damage. Permeation of reduced, reactive iron (Fe2+) through LTCCs in iron overload may help explain the profound contractile dysfunction, Ca2+ overload and impaired diastolic function commonly observed during the early stages of iron overload [13, 42, 84, 88]. Specifically, the reduced iron entering through LTCCs has preferential access to the major regulators of excitation–contraction coupling (Fig. 2). Moreover, Fe2+-induced slowing of LTCC current inactivation by Fe2+ leads to increased Ca2+ entry and Ca2+ overload [68]. Since elevated Ca2+, particularly through LTCCs, is involved in the initiation and progression of heart disease in a calcineurin-dependent manner [144], it is conceivable that the slowing of LTCC inactivation by NTBI may contribute to the cardiomyopathy seen in iron overload. Additionally, slowed inactivation of LTCCs positively feedback to further increase NTBI Fe2+ entry into cardiomyocytes, thereby amplifying alterations in cellular function. Elevations in cytosolic iron also lead to iron uptake into mitochondria where it normally becomes incorporated into Fe–S clusters and is utilized for heme biosynthesis [3]. Iron homeostasis in the mitochondria requires frataxin, a ferroxidase that detoxifies redox-active iron, thereby providing a critical anti-oxidative defense against iron-derived radicals in the mitochondria [145, 146]. Frataxin deficiency causes a clinical syndrome called Friedreich ataxia that involves neurodegeneration, dilated cardiomyopathy and diabetes mellitus [147].
Although our previous results provide unequivocal evidence for NTBI transport by LTCC in heart, the application of LTCC blockers failed to alter iron uptake rates in cultured rat neonatal myoctes [64]. However, these studies should be interpreted with caution for several reasons. First, neonatal myocytes have much smaller L-type Ca2+ current compared to adult myocardium [148]. Second, when culturing neonatal cardiomyocytes at low densities, the fraction of beating myocytes is very low and beating rates are slow compared to normal heart rates. Therefore, it is imperative that electrical pacing be used in conjunction with high-density cultures when examining the role of LTCC in iron transport. Third, neonatal cultures suffer from high rates of myocytes death, particularly in the presence of iron, making it essential to consider the number of viable cells in the calculation of iron uptake rates. Fourth, our results predict that LTCCs also contribute to ferrous iron export. Consequently, at non-physiologically high iron loads (as used in the cultured myocyte studies), LTCC blockers are predicted to have a limited impact on the net iron uptake. Collectively, the validity of the neonatal cultured myocyte system as a tool to test the role of LTCCs as iron transporters is questionable [64].
Endocrine dysfunction in patients with iron-overload: a potential role for LTCC
L-type Ca2+ channels are present in large numbers in pancreatic beta cells [127, 128] and in various cell types of the anterior pituitary gland including gonadotrophs [149–151], thyrotrophs [152], corticoptrophs [153] and in the parathyroid-hormone-producing cells of the parathyroid gland (Table 3) [154]. In these endocrine cells, LTCCs are critically involved in excitation–secretion coupling whereby membrane depolarization activate (open) LTCCs, allowing extracellular Ca2+ entry and stimulation of exocytosis [127, 149, 151]. Given the important role of LTCC in mediating cellular NTBI uptake, it seems plausible to suggest a link between LTCCs and iron-overload-induced dysfunction in these endocrine tissues. Similar to cardiomyocytes, excessive Fe2+ entry through LTCCs is expected to impair secretion and increase cell loss (see Table 3). Consistent with this mechanism, histological and immunohistochemical analyses have confirmed deposition of iron in human beta cells [155], while imaging studies have shown iron accumulation in the anterior pituitary gland in patients with iron overload [156].
These observations help explain the observation that endocrinopathies are common co-morbidities present in patients with iron overload and include diabetes mellitus [21, 22, 24, 155], hypoparathyroidism [22, 157] and anterior pituitary dysfunction associated with hypopituitary hypogonadism [22–24], impaired adrenocorticotrophin reserve [21, 23] and hypothyroidism [22]. Diabetes mellitus in patients with iron overload is associated with reduced insulin reserve and impaired insulin release. Consequently, most patients often progress to be insulin-dependent relatively early during the course of illness [21, 22, 158]. In addition, up to 50% of patients with beta-thalassemia major have a short stature and failure of puberty associated with primary and secondary amenorrhea in women [22, 24].
Therapeutic implications for CCBs under iron-overload conditions
Calcium channel blockers (CCBs) are widely used to treat cardiovascular diseases in both pediatric [159] and adult [160, 161] populations. The recognition that LTCCs are critical transporters of iron under conditions of iron overload opens up the possibility of using CCBs in the treatment and/or prevention of iron-overload cardiomyopathy in combination with standard chelation therapy. Unlike iron chelators that are effective in removing excess iron accumulation in cells, CCBs are expected to be particularly effective early in iron overload by reducing tissue iron uptake and preventing disease progression. In advanced iron overload, the potential effectiveness of CCBs may be limited, but they should block further iron uptake, thereby enhancing the effectiveness of iron chelation therapies. However, the applicability of CCBs in the setting of advanced dilated cardiomyopathy or conduction block may be limited due to their negative inotropic and chronotropic effects.
While the data supporting the potential effectiveness of CCBs in the treatment of iron-overload cardiomypathy has been obtained in rodents, pharmacological blockade of LTCC might have an even greater therapeutic benefit in humans for two reasons: (a) humans lack iron excretory pathways that are present in rodents [1, 2], and (b) humans have proportionately larger trans-sarcolemmal I Ca,L influx per cardiac cycle [162]. The translation of the rodent studies to humans is bolstered by the observation that the plasma concentration of CCBs required to reduce iron accumulation in murine hearts [18] is similar to levels reported in patients [163–165]. Blockade of the LTCC may also provide additional benefits beyond a direct inhibition of Fe2+ entry into cardiomyocytes and endocrine tissues. For example, CCBs could potentially reduce Fe2+-induced Ca2+ overload as a result of LTCC inactivation [68] and thereby facilitate diastolic ventricular filling [166]. Finally, calcium channel blockers may promote myocardial microvascular perfusion by vasodilating coronary arterioles while improving coronary endothelial function [132, 167]. LTCC blockers like amlodipine also possess antioxidant properties that could help offset the oxidative effects of iron overload [167]. Given the high degree of oxidative stress, diastolic dysfunction and possibility for coronary endothelial dysfunction in iron-overload cardiomyopathy, these effects may increase the potential therapeutic benefits of CCBs in patients with iron overload. In addition to protecting the heart, pharmacological blockade of LTCCs may also reduce the endocrinopathies observed in iron-overload patients. Clearly, clinical trials are warranted to establish the clinical efficacy and safety of CCB treatment in patients with iron overload.
Abbreviations
- LTCC:
-
L-type calcium channel
- I Ca :
-
LTCC current
- NTBI:
-
Non-transferrin bound iron
- CCB:
-
Calcium channel blocker
- AV:
-
Atrioventricular
- SA:
-
Sinoatrial
- VSMCs:
-
Vascular smooth muscle cells
- LIP:
-
Labile intracellular iron pool
- DMT1:
-
Divalent metal transporter 1
- TfR:
-
Transferrin receptor
References
Andrews NC (1999) Disorders of iron metabolism. N Engl J Med 341:1986–1995
Hentze MW, Muckenthaler MU, Andrews NC (2004) Balancing acts; molecular control of mammalian iron metabolism. Cell 117:285–297
Napier I, Ponka P, Richardson DR (2005) Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood 105:1867–1874
Weatherall DJ, Clegg JB (1996) Thalassemia—a global public health problem. Nat Med 2:847–849
Olivieri NF (1999) The beta-thalassemias. N Engl J Med 341:99–109
Weatherall DJ, Clegg JB (2001) Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ 79:704–712
Pietrangelo A (2004) Hereditary hemochromatosis—a new look at an old disease. N Engl J Med 350:2383–2397
Breuer W, Hershko C, Cabantchik ZI (2000) The importance of non-transferrin bound iron in disorders of iron metabolism. Transfus Sci 23:185–192
Templeton DM, Liu Y (2003) Genetic regulation of cell function in response to iron overload or chelation. Biochim Biophys Acta 1619:113–124
Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C, Cabantchik ZI (2003) Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood 102:2670–3677
Kaplan J, Jordan I, Sturrock A (1991) Regulation of the transferrin-independent iron transport system in cultured cells. J Biol Chem 266:2997–3004
Randell EW, Parkes JG, Olivieri NF, Templeton DM (1994) Uptake of non-transferrin-bound iron by both reductive and nonreductive processes is modulated by intracellular iron. J Biol Chem 269:16046–16053
Liu P, Olivieri N (1994) Iron overload cardiomyopathies: new insights into an old disease. Cardiovasc Drugs Ther 8:101–110
Lee DH, Jacobs DR Jr (2004) Serum markers of stored body iron are not appropriate markers of health effects of iron: a focus on serum ferritin. Med Hypotheses 62:442–445
Gutteridge JM, Rowley DA, Griffiths E, Halliwell B (1985) Low-molecular-weight iron complexes and oxygen radical reactions in idiopathic haemochromatosis. Clin Sci (Colch) 68:463–467
Artman M, Olson RD, Boucek RJ Jr, Boerth RC (1984) Depression of contractility in isolated rabbit myocardium following exposure to iron: role of free radicals. Toxicol Appl Pharmacol 72:324–332
Vernon DD, Banner W Jr, Dean JM (1989) Hemodynamic effects of experimental iron poisoning. Ann Emerg Med 18:863–866
Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP, Backx PH (2003) L-type Ca(2+) channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med 9:1187–1194
Oudit GY, Trivieri MG, Khaper N, Husain T, Wilson GJ, Liu P, Sole MJ, Backx PH (2004) Taurine supplementation reduces oxidative stress and improves cardiovascular function in an iron-overload murine model. Circulation 109:1877–1885
Seznec H, Simon D, Monassier L, Criqui-Filipe P, Gansmuller A, Rustin P, Koenig M, Puccio H (2004) Idebenone delays the onset of cardiac functional alteration without correction of Fe–S enzymes deficit in a mouse model for Friedreich ataxia. Hum Mol Genet 13:1017–1024
Schafer AI, Cheron RG, Dluhy R, Cooper B, Gleason RE, Soeldner JS, Bunn HF (1981) Clinical consequences of acquired transfusional iron overload in adults. N Engl J Med 304:319–324
Italian Working Group (1995) Multicentre study on prevalence of endocrine complications in thalassaemia major. Italian Working Group on Endocrine Complications in Non-endocrine Diseases. Clin Endocrinol (Oxf) 42:581–586
Hempenius LM, Van Dam PS, Marx JJ, Koppeschaar HP (1999) Mineralocorticoid status and endocrine dysfunction in severe hemochromatosis. J Endocrinol Invest 22:369–376
Li CK, Luk CW, Ling SC, Chik KW, Yuen HL, Shing MM, Chang KO, Yuen PM (2002) Morbidity and mortality patterns of thalassaemia major patients in Hong Kong: retrospective study. Hong Kong Med J 8:255–260
Sinigaglia L, Fargion S, Fracanzani AL, Binelli L, Battafarano N, Varenna M, Piperno A, Fiorelli G (1997) Bone and joint involvement in genetic hemochromatosis: role of cirrhosis and iron overload. J Rheumatol 24:1809–1813
Matsushima S, Torii M, Ozaki K, Narama I (2003) Iron lactate-induced osteomalacia in association with osteoblast dynamics. Toxicol Pathol 31:646–654
Mahachoklertwattana P, Sirikulchayanonta V, Chuansumrit A, Karnsombat P, Choubtum L, Sriphrapradang A, Domrongkitchaiporn S, Sirisriro R, Rajatanavin R (2003) Bone histomorphometry in children and adolescents with beta-thalassemia disease: iron-associated focal osteomalacia. J Clin Endocrinol Metab 88:3966–3972
Voest EE, Vreugdenhil G, Marx JJ (1994) Iron-chelating agents in non-iron overload conditions. Ann Intern Med 120:490–499
Turoczi T, Jun L, Cordis G, Morris JE, Maulik N, Stevens RG, Das DK (2003) HFE mutation and dietary iron content interact to increase ischemia/reperfusion injury of the heart in mice. Circ Res 92:1240–1246
de Valk B, Marx JJ (1999) Iron, atherosclerosis, and ischemic heart disease. Arch Intern Med 159:1542–1548
Ramakrishna G, Rooke TW, Cooper LT (2003) Iron and peripheral arterial disease: revisiting the iron hypothesis in a different light. Vasc Med 8:203–210
Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK (2003) Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron 37:899–909
Todorich BM, Connor JR (2004) Redox metals in Alzheimer's disease. Ann N Y Acad Sci 1012:171–178
Rochette J, Pointon JJ, Fisher CA, Perera G, Arambepola M, Arichchi DS, De Silva S, Vandwalle JL, Monti JP, Old JM, Merryweather-Clarke AT, Weatherall DJ, Robson KJ (1999) Multicentric origin of hemochromatosis gene (HFE) mutations. Am J Hum Genet 64:1056–1062
Bulaj ZJ, Ajioka RS, Phillips JD, LaSalle BA, Jorde LB, Griffen LM, Edwards CQ, Kushner JP (2000) Disease-related conditions in relatives of patients with hemochromatosis. N Engl J Med 343:1529–1535
Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR, Goldberg YP (2004) Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet 36:77–82
Roetto A, Daraio F, Alberti F, Porporato P, Cali A, De Gobbi M, Camaschella C (2002) Hemochromatosis due to mutations in transferrin receptor 2. Blood Cells Mol Dis 29:465–470
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–2093
De Domenico I, Ward DM, Nemeth E, Vaughn MB, Musci G, Ganz T, Kaplan J (2005) The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci U S A 102:8955–8960
Gordeuk VR (2002) African iron overload. Semin Hematol 39:263–269
Loreal O, Gosriwatana I, Guyader D, Porter J, Brissot P, Hider RC (2000) Determination of non-transferrin-bound iron in genetic hemochromatosis using a new HPLC-based method. J Hepatol 32:727–733
Chen FE, Ooi C, Ha SY, Cheung BM, Todd D, Liang R, Chan TK, Chan V (2000) Genetic and clinical features of hemoglobin H disease in Chinese patients. N Engl J Med 343:544–550
Olivieri NF (2001) Progression of iron overload in sickle cell disease. Semin Hematol 38:57–62
Bottomley SS (2001) Iron overload in sideroblastic and other non-thalassemic anemias. In: Barton JC, Edwards CQ (eds) Hemochromatosis: genetics, pathophysiology, diagnosis, and treatment. Cambridge University Press, Cambridge, pp 442–467
Quinn CT, Rogers ZR, Buchanan GR (2004) Survival of children with sickle cell disease. Blood 103:4023–4027
Steinberg MH (1999) Management of sickle cell disease. N Engl J Med 340:1021–1030
Serjeant GR, Serjeant BE (2001) Sickle cell disease, 3rd edn. Oxford University Press, New York
Fleming MD (2002) The genetics of inherited sideroblastic anemias. Semin Hematol 39:270–281
Zager RA, Johnson AC, Hanson SY, Wasse H (2002) Parenteral iron formulations: a comparative toxicologic analysis and mechanisms of cell injury. Am J Kidney Dis 40:90–103
Kalantar-Zadeh K, Don BR, Rodriguez RA, Humphreys MH (2001) Serum ferritin is a marker of morbidity and mortality in hemodialysis patients. Am J Kidney Dis 37:564–572
Kletzmayr J, Horl WH (2002) Iron overload and cardiovascular complications in dialysis patients. Nephrol Dial Transplant 17(Suppl 2):25–29
Engle JP, Polin KS, Stile IL (1987) Acute iron intoxication: treatment controversies. Drug Intell Clin Pharm 21:153–159
Tenenbein M, Kopelow ML, deSa DJ (1988) Myocardial failure and shock in iron poisoning. Human Toxicol 7:281–284
Fine JS (2000) Iron poisoning. Curr Probl Pediatr 30:71–90
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 21:195–199
Robb A, Wessling-Resnick M (2004) Regulation of transferrin receptor 2 protein levels by transferrin. Blood 104:4294–4299
Johnson MB, Enns CA (2004) Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 104:4287–4293
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482–488
Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, Hentze MW, Rouault TA, Andrews NC, Hediger MA (2001) Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett 509:309–316
Ke Y, Chen YY, Chang YZ, Duan XL, Ho KP, Jiang de H, Wang K, Qian ZM (2003) Post-transcriptional expression of DMT1 in the heart of rat. J Cell Physiol 196:124–130
Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL (2004) Identification of a human heme exporter that is essential for erythropoiesis. Cell 118:757–766
Thomas C, Oates PS (2004) Ferroportin/IREG-1/MTP-1/SLC40A1 modulates the uptake of iron at the apical membrane of enterocytes. Gut 53:44–49
Scheiber-Mojdehkar B, Lutzky B, Schaufler R, Sturm B, Goldenberg H (2004) Non-transferrin-bound iron in the serum of hemodialysis patients who receive ferric saccharate: no corretion to peroxide generation. J Am Soc Nephrol 15:1648–1655
Parkes JG, Olivieri NF, Templeton DM (1997) Characterization of Fe2+ and Fe3+ transport by iron-loaded cardiac myocytes. Toxicology 117:141–151
Lansman JB, Hess P, Tsien RW (1986) Blockade of current through single calcium channels by Cd2+, Mg2+, and Ca2+. Voltage and concentration dependence of calcium entry into the pore. J Gen Physiol 88:321–347
Winegar BD, Kelly R, Lansman JB (1991) Block of current through single calcium channels by Fe, Co, and Ni. Location of the transition metal binding site in the pore. J Gen Physiol 97:351–367
Atar D, Backx PH, Appel MM, Gao WD, Marban E (1995) Excitation–transcription coupling mediated by zinc influx through voltage-dependent calcium channels. J Biol Chem 270:2473–2477
Tsushima RG, Wickenden AD, Bouchard RA, Oudit GY, Liu PP, Backx PH (1999) Modulation of iron uptake in heart by L-type Ca2+ channel modifiers: possible implications in iron overload. Circ Res 84:1302–1309
Townsend A, Drakesmith H (2002) Role of HFE in iron metabolism, hereditary haemochromatosis, anaemia of chronic disease, and secondary iron overload. Lancet 359:786–790
Laftah AH, Ramesh B, Simpson RJ, Solanky N, Bahram S, Schumann K, Debnam ES, Srai SK (2004) Effect of hepcidin on intestinal iron absorption in mice. Blood 103:3940–3944
Ludwiczek S, Theurl I, Bahram S, Schumann K, Weiss G (2005) Regulatory networks for the control of body iron homeostasis and their dysregulation in HFE mediated hemochromatosis. J Cell Physiol 204:489–499
Yamaji S, Sharp P, Ramesh B, Srai SK (2004) Inhibition of iron transport across human intestinal epithelial cells by hepcidin. Blood 104:2178–2180
Muckenthaler M, Roy CN, Custodio AO, Minana B, deGraaf J, Montross LK, Andrews NC, Hentze MW (2003) Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat Genet 34:102–107
Celec P (2005) Hemojuvelin: a supposed role in iron metabolism one year after its discovery. J Mol Med 83:521–525
Niederkofler V, Salie R, Arber S (2005) Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest 115:2180–2186
Ward RJ, Kuhn LC, Kaldy P, Florence A, Peters TJ, Crichton RR (1994) Control of cellular iron homeostasis by iron-responsive elements in vivo. Eur J Biochem 220:927–931
Pantopoulos K (2004) Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci 1012:1–13
Meyron-Holtz EG, Ghosh MC, Rouault TA (2004) Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science 306:2087–2090
Olivieri NF, Nathan DG, MacMillan JH, Wayne AS, Liu PP, McGee A, Martin M, Koren G, Cohen AR (1994) Survival in medically treated patients with homozygous beta-thalassemia. N Engl J Med 331:574–578
Brittenham GM, Griffith PM, Nienhuis AW, McLaren CE, Young NS, Tucker EE, Allen CJ, Farrell DE, Harris JW (1994) Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N Engl J Med 331:567–573
Zurlo MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, Melevendi C, Di Gregorio F, Burattini MG, Terzoli S (1989) Survival and causes of death in thalassaemia major. Lancet 2:27–30
Niederau C, Fischer R, Purschel A, Stremmel W, Haussinger D, Strohmeyer G (1996) Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 110:1107–1119
Cecchetti G, Binda A, Piperno A, Nador F, Fargion S, Fiorelli G (1991) Cardiac alterations in 36 consecutive patients with idiopathic haemochromatosis: polygraphic and echocardiographic evaluation. Eur Heart J 12:224–230
Muhlestein JB (2000) Cardiac abnormalities in hemochromatosis. In: Barton JC, Edwards CQ (eds) Hemochromatosis: genetics, pathophysiology, diagnosis, and treatment. Cambridge University Press, Cambridge, pp 297–310
Palka P, Macdonald G, Lange A, Burstow DJ (2002) The role of Doppler left ventricular filling indexes and Doppler tissue echocardiography in the assessment of cardiac involvement in hereditary hemochromatosis. J Am Soc Echocardiogr 15:884–890
Modell B, Khan M, Darlison M (2000) Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet 355:2051–2052
Veglio F, Melchio R, Rabbia F, Molino P, Genova GC, Martini G, Schiavone D, Piga A, Chiandussi L (1998) Blood pressure and heart rate in young thalassemia major patients. Am J Hypertens 11:539–547
Kremastinos DT, Tsiapras DP, Tsetsos GA, Rentoukas EI, Vretou HP, Toutouzas PK (1993) Left ventricular diastolic Doppler characteristics in beta-thalassemia major. Circulation 88:1127–1135
Buja LM, Roberts WC (1971) Iron in the heart. Etiology and clinical significance. Am J Med 51:209–221
Mattheyses M, Hespel JP, Brissot P, Daubert JC, Hita de Nercy Y, Lancien G, Le Treut A, Pony JC, Simon M, Ferrand B, Gouffault J, Bourel M (1978) The cardiomyopathy of idiopathic hemochromatosis. Arch Mal Coeur Vaiss 71:371–379
Rosenqvist M, Hultcrantz R (1989) Prevalence of a haemochromatosis among men with clinically significant bradyarrhythmias. Eur Heart J 10:473–478
Laurita KR, Chuck ET, Yang T, Dong WQ, Kuryshev YA, Brittenham GM, Rosenbaum DS, Brown AM (2003) Optical mapping reveals conduction slowing and impulse block in iron-overload cardiomyopathy. J Lab Clin Med 142:83–89
Eaton JW, Qian M (2002) Molecular bases of cellular iron toxicity. Free Radic Biol Med 32:833–840
Kadiiska MB, Burkitt MJ, Xiang QH, Mason RP (1995) Iron supplementation generates hydroxyl radical in vivo. An ESR spin-trapping investigation. J Clin Invest 96:1653–1657
Young IS, Trouton TG, Torney JJ, McMaster D, Callender ME, Trimble ER (1994) Antioxidant status and lipid peroxidation in hereditary haemochromatosis. Free Radic Biol Med 16:393–397
Livrea MA, Tesoriere L, Pintaudi AM, Calabrese A, Maggio A, Freisleben HJ, D'Arpa D, D'Anna R, Bongiorno A (1996) Oxidative stress and antioxidant status in beta-thalassemia major: iron overload and depletion of lipid-soluble antioxidants. Blood 88:3608–3614
Lim PS, Chan EC, Lu TC, Yu YL, Kuo SY, Wang TH, Wei YH (2000) Lipophilic antioxidants and iron status in ESRD patients on hemodialysis. Nephron 86:428–435
Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11:81–128
Lee SH, Oe T, Blair IA (2001) Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science 292:2083–2086
Valenti L, Conte D, Piperno A, Dongiovanni P, Fracanzani AL, Fraquelli M, Vergani A, Gianni C, Carmagnola L, Fargion S (2004) The mitochondrial superoxide dismutase A16V polymorphism in the cardiomyopathy associated with hereditary haemochromatosis. J Med Genet 41:946–950
Kremastinos DT, Tiniakos G, Theodorakis GN, Katritsis DG, Toutouzas PK (1995) Myocarditis in beta-thalassemia major. A cause of heart failure. Circulation 91:66–71
Kremastinos DT, Flevari P, Spyropoulou M, Vrettou H, Tsiapras D, Stavropoulos-Giokas CG (1999) Association of heart failure in homozygous beta-thalassemia with the major histocompatibility complex. Circulation 100:2074–2078
Economou-Petersen E, Aessopos A, Kladi A, Flevari P, Karabatsos F, Fragodimitri C, Nicolaidis P, Vrettou H, Vassilopoulos D, Karagiorga-Lagana M, Kremastinos DT, Petersen MB (1998) Apolipoprotein E epsilon4 allele as a genetic risk factor for left ventricular failure in homozygous beta-thalassemia. Blood 92:3455–3459
Cooper JM, Schapira AH (2003) Friedreich's ataxia: disease mechanisms, antioxidant and coenzyme Q10 therapy. Biofactors 18:163–171
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662
Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16:521–555
Mukherjee R, Spinale FG (1998) L-type calcium channel abundance and function with cardiac hypertrophy and failure: a review. J Mol Cell Cardiol 30:1899–1916
Bers DM (2002) Excitation–contraction coupling and cardiac contractile force, 2nd edn. Kluwer, Dordrecht, pp 101–132
Cherednichenko G, Zima AV, Feng W, Schaefer S, Blatter LA, Pessah IN (2004) NADH oxidase activity of rat cardiac sarcoplasmic reticulum regulates calcium-induced calcium release. Circ Res 94:478–486
Goldhaber JI, Qayyum MS (2000) Oxygen free radicals and excitation–contraction coupling. Antioxid Redox Signal 2:55–64
Lacampagne A, Duittoz A, Bolanos P, Peineau N, Argibay JA (1995) Effect of sulfhydryl oxidation on ionic and gating currents associated with L-type calcium channels in isolated guinea-pig ventricular myocytes. Cardiovasc Res 30:799–806
Shirotani K, Katsura M, Higo A, Takesue M, Mohri Y, Shuto K, Tarumi C, Ohkuma S (2001) Suppression of Ca2+ influx through L-type voltage-dependent calcium channels by hydroxyl radical in mouse cerebral cortical neurons. Brain Res Mol Brain Res 92:12–18
Kuryshev YA, Brittenham GM, Fujioka H, Kannan P, Shieh CC, Cohen SA, Brown AM (1999) Decreased sodium and increased transient outward potassium currents in iron-loaded cardiac myocytes. Implications for the arrhythmogenesis of human siderotic heart disease. Circulation 100:675–683
Horackova M, Ponka P, Byczko Z (2000) The antioxidant effects of a novel iron chelator salicylaldehyde isonicotinoyl hydrazone in the prevention of H(2)O(2) injury in adult cardiomyocytes. Cardiovasc Res 47:529–536
Folden DV, Gupta A, Sharma AC, Li SY, Saari JT, Ren J (2003) Malondialdehyde inhibits cardiac contractile function in ventricular myocytes via a p38 mitogen-activated protein kinase-dependent mechanism. Br J Pharmacol 139:1310–1316
Schwartz KA, Li Z, Schwartz DE, Cooper TG, Braselton WE (2002) Earliest cardiac toxicity induced by iron overload selectively inhibits electrical conduction. J Appl Physiol 93:746–751
Tomaselli GF, Zipes DP (2004) What causes sudden death in heart failure? Circ Res 95:754–763
Barrington PL, Martin RL, Zhang K (1997) Slowly inactivating sodium currents are reduced by exposure to oxidative stress. J Mol Cell Cardiol 29:3251–3265
Wu VC, Huang JW, Wu MS, Chin CY, Chiang FT, Liu YB, Wu KD (2004) The effect of iron stores on corrected QT dispersion in patients undergoing peritoneal dialysis. Am J Kidney Dis 44:720–728
Gaenzer H, Marschang P, Sturm W, Neumayr G, Vogel W, Patsch J, Weiss G (2002) Association between increased iron stores and impaired endothelial function in patients with hereditary hemochromatosis. J Am Coll Cardiol 40:2189–2194
Cheung YF, Chan GC, Ha SY (2002) Arterial stiffness and endothelial function in patients with beta-thalassemia major. Circulation 106:2561–2566
Lemogoum D, Van Bortel L, Najem B, Dzudie A, Teutcha C, Madu E, Leeman M, Degaute JP, van de Borne P (2004) Arterial stiffness and wave reflections in patients with sickle cell disease. Hypertension 44:924–929
Liu Y, Parkes JG, Templeton DM (2003) Differential accumulation of non-transferrin-bound iron by cardiac myocytes and fibroblasts. J Mol Cell Cardiol 35:505–514
Hess P, Lansman JB, Tsien RW (1986) Calcium channel selectivity for divalent and monovalent cations. Voltage and concentration dependence of single channel current in ventricular heart cells. J Gen Physiol 88:293–319
Rychkov G, Brereton HM, Harland ML, Barritt GJ (2001) Plasma membrane Ca2+ release-activated Ca2+ channels with a high selectivity for Ca2+ identified by patch-clamp recording in rat liver cells. Hepatology 33:938–947
Jorgensen NR, Teilmann SC, Henriksen Z, Civitelli R, Sorensen OH, Steinberg TH (2003) Activation of L-type calcium channels is required for gap junction-mediated intercellular calcium signaling in osteoblastic cells. J Biol Chem 278:4082–4086
Rorsman P, Renstrom E (2003) Insulin granule dynamics in pancreatic beta cells. Diabetologia 46:1029–1045
Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig JJ (2004) Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca 2+ channels. J Clin Invest 113:1430–1439
Van Goor F, Zivadinovic D, Stojilkovic SS (2001) Differential expression of ionic channels in rat anterior pituitary cells. Mol Endocrinol 15:1222–1236
Tsien RW, Hess P, McCleskey EW, Rosenberg RL (1987) Calcium channels: mechanisms of selectivity, permeation, and block. Annu Rev Biophys Biophys Chem 16:265–290
Cataldi M, Perez-Reyes E, Tsien RW (2002) Differences in apparent pore sizes of low and high voltage-activated Ca2+ channels. J Biol Chem 277:45969–45976
Abernethy DR, Schwartz JB (1999) Calcium-antagonist drugs. N Engl J Med 341:1447–1457
Lipscombe D (2002) L-type calcium channels: highs and new lows. Circ Res 90:933–935
Han W, Chartier D, Li D, Nattel S (2001) Ionic remodeling of cardiac Purkinje cells by congestive heart failure. Circulation 104:2095–2100
Biel M, Schneider A, Wahl C (2002) Cardiac HCN channels: structure, function, and modulation. Trends Cardiovasc Med 12:206–212
Mitcheson JS, Hancox JC (1997) Modulation by mexiletine of action potentials, L-type Ca current and delayed rectifier K current recorded from isolated rabbit atrioventricular nodal myocytes. Pflugers Arch 434:855–858
Verkerk AO, Wilders R, Coronel R, Ravesloot JH, Verheijck EE (2003) Ionic remodeling of sinoatrial node cells by heart failure. Circulation 108:760–766
Fassos FF, Berkovitch M, Daneman N, Koren L, Cameron R, Klein J, Falcitelli C, St Louis P, Daneman R, Koren G (1998) The efficacy of diazepam in the treatment of acute iron overload in rats. Can J Physiol Pharmacol 76:895–899
Hara Y, Kobayashi H, Ooshiro S, Futamura K, Nishino T, Chugun A, Temma K, Kondo H (2001) Negative inotropic effect of diazepam in isolated guinea pig heart. J Vet Med Sci 63:135–143
Knabb RM, Rosamond TL, Fox KA, Sobel BE, Bergmann SR (1986) Enhancement of salvage of reperfused ischemic myocardium by diltiazem. J Am Coll Cardiol 8:861–871
Ehring T, Heusch G (1995) Stunned myocardium and the attenuation of stunning by calcium antagonists. Am J Cardiol 75:61E–67E
Horwitz LD, Rosenthal EA (1999) Iron-mediated cardiovascular injury. Vasc Med 4:93–99
Murray MT, White K, Munro HN (1987) Conservation of ferritin heavy subunit gene structure: implications for the regulation of ferritin gene expression. Proc Natl Acad Sci U S A 84:7438–7442
Sah R, Oudit GY, Nguyen TT, Lim HW, Wickenden AD, Wilson GJ, Molkentin JD, Backx PH (2002) Inhibition of calcineurin and sarcolemmal Ca2+ influx protects cardiac morphology and ventricular function in K(v)4.2N transgenic mice. Circulation 105:1850–1856
O'Neill HA, Gakh O, Park S, Cui J, Mooney SM, Sampson M, Ferreira GC, Isaya G (2005) Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry 44:537–545
Sturm B, Bistrich U, Schranzhofer M, Sarsero JP, Rauen U, Scheiber-Mojdehkar B, de Groot H, Ioannou P, Petrat F (2005) Friedreich's ataxia, no changes in mitochondrial labile iron in human lymphoblasts and fibroblasts: a decrease in antioxidative capacity? J Biol Chem 280:6701–6708
Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276:1709–1712
Liu L, O'Hara DS, Cala SE, Poornima I, Hines RN, Marsh JD (2000) Developmental regulation of the L-type calcium channel alpha1C subunit expression in heart. Mol Cell Biochem 205:101–109
Hezareh M, Schlegel W, Rawlings SR (1997) Stimulation of Ca2+ influx in alpha T3-1 gonadotrophs via the cAMP/PKA signaling system. Am J Physiol 273:E850–E858
Anderson L, Hoyland J, Mason WT, Eidne KA (1992) Characterization of the gonadotrophin-releasing hormone calcium response in single alpha T3-1 pituitary gonadotroph cells. Mol Cell Endocrinol 86:167–175
Mulvaney JM, Zhang T, Fewtrell C, Roberson MS (1999) Calcium influx through L-type channels is required for selective activation of extracellular signal-regulated kinase by gonadotropin-releasing hormone. J Biol Chem 274:29796–29804
Shupnik MA, Weck J, Hinkle PM (1996) Thyrotropin (TSH)-releasing hormone stimulates TSH beta promoter activity by two distinct mechanisms involving calcium influx through L type Ca2+ channels and protein kinase C. Mol Endocrinol 10:90–99
Fiekers JF, Konopka LM (1996) Spontaneous transients of [Ca2+]i depend on external calcium and the activation of L-type voltage-gated calcium channels in a clonal pituitary cell line (AtT-20) of cultured mouse corticotropes. Cell Calcium 19:327–336
Chang W, Pratt SA, Chen TH, Tu CL, Mikala G, Schwartz A, Shoback D (2001) Parathyroid cells express dihydropyridine-sensitive cation currents and L-type calcium channel subunits. Am J Physiol Endocrinol Metab 281:E180–E189
Rahier J, Loozen S, Goebbels RM, Abrahem M (1987) The haemochromatotic human pancreas: a quantitative immunohistochemical and ultrastructural study. Diabetologia 30:5–12
Argyropoulou MI, Kiortsis DN, Metafratzi Z, Bitsis S, Tsatoulis A, Efremidis SC (2001) Pituitary gland height evaluated by MR in patients with beta-thalassemia major: a marker of pituitary gland function. Neuroradiology 43:1056–1058
Economou M, Katzos G, Koussi A, Tsatra I, Athanassiou-Metaxa M (2003) Hypoparathyroidism in beta-thalassemic patients. J Pediatr Hematol Oncol 25:275–276 (author reply 276)
Cario H, Holl RW, Debatin KM, Kohne E (2003) Insulin sensitivity and beta-cell secretion in thalassaemia major with secondary haemochromatosis: assessment by oral glucose tolerance test. Eur J Pediatr 162:139–146
Flynn JT, Pasko DA (2000) Calcium channel blockers: pharmacology and place in therapy of pediatric hypertension. Pediatr Nephrol 15:302–316
Pepine CJ, Handberg EM, Cooper-DeHoff RM, Marks RG, Kowey P, Messerli FH, Mancia G, Cangiano JL, Garcia-Barreto D, Keltai M, Erdine S, Bristol HA, Kolb HR, Bakris GL, Cohen JD, Parmley WW (2003) A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil–Trandolapril Study (INVEST): a randomized controlled trial. JAMA 290:2805–2816
Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, Hansson L, Hua T, Laragh J, McInnes GT, Mitchell L, Plat F, Schork A, Smith B, Zanchetti A (2004) Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet 363:2022–2031
Bers DM (2002) Cardiac excitation–contraction coupling. Nature 415:198–205
Mikus G, Eichelbaum M, Fischer C, Gumulka S, Klotz U, Kroemer HK (1990) Interaction of verapamil and cimetidine: stereochemical aspects of drug metabolism, drug disposition and drug action. J Pharmacol Exp Ther 253:1042–1048
Fuhr U, Muller-Peltzer H, Kern R, Lopez-Rojas P, Junemann M, Harder S, Staib AH (2002) Effects of grapefruit juice and smoking on verapamil concentrations in steady state. Eur J Clin Pharmacol 58:45–53
Elliott HL, Elawad M, Wilkinson R, Singh SP (2002) Persistence of antihypertensive efficacy after missed doses: comparison of amlodipine and nifedipine gastrointestinal therapeutic system. J Hypertens 20:333–338
Murphy SW (2004) Diastolic dysfunction. Curr Treat Options Cardiovasc Med 6:61–68
Mason RP, Marche P, Hintze TH (2003) Novel vascular biology of third-generation L-type calcium channel antagonists: ancillary actions of amlodipine. Arterioscler Thromb Vasc Biol 23:2155–2163
Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Jr., Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Wolff RK, et al (1996) A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 13:399–408
Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC (1999) The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood 94:9–11
Roetto A, Daraio F, Porporato P, Caruso R, Cox TM, Cazzola M, Gasparini P, Piperno A, Camaschella C (2003) Screening hepcidin for mutations in juvenile hemochromatosis: identification of a new mutation (C70r). Blood
Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, Majorano N, Totaro A, Gasparini P (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 25:14–15
Fleming RE, Ahmann JR, Migas MC, Waheed A, Koeffler HP, Kawabata H, Britton RS, Bacon BR, Sly WS (2002) Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc Natl Acad Sci U S A 99:10653–10658
Njajou OT, Vaessen N, Joosse M, Berghuis B, van Dongen JW, Breuning MH, Snijders PJ, Rutten WP, Sandkuijl LA, Oostra BA, van Duijn CM, Heutink P (2001) A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet 28:213–214
Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, Trenor CC, Gasparini P, Andrews NC, Pietrangelo A (2001) Autosomaldominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 108:619–623
Shehee WR, Oliver P, Smithies O (1993) Lethal thalassemia after insertional disruption of the mouse major adult beta-globin gene. Proc Natl Acad Sci U S A 90:3177–3181
Yang B, Kirby S, Lewis J, Detloff P, Maeda N, Smithies O (1995) A Mouse Model for {beta}0-Thalassemia. PNAS 92:11608–11612
Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, Rubin EM (1997) Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science 278:876–878
Ryan TM, Ciavatta DJ, Townes TM (1997) Knockout-transgenic mouse model of sickle cell disease. Science 278:873–876
Yamamoto M, Nakajima O (2000) Animal models for X-linked sideroblastic anemia. Int J Hematol 72:157–164
Nakajima O, Takahashi S, Harigae H, Furuyama K, Hayashi N, Sassa S, Yamamoto M (1999) Heme deficiency in erythroid lineage causes differentiation arrest and cytoplasmic iron overload. EMBO J 18:6282–6289
Heimpel H, Anselstetter V, Chrobak L, Denecke J, Einsiedler B, Gallmeier K, Griesshammer A, Marquardt T, Janka-Schaub G, Kron M, Kohne E (2003) Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long-term observation. Blood 102:4576–4581
Lim PS, Wei YH, Yu YL, Kho B (1999) Enhanced oxidative stress in haemodialysis patients receiving intravenous iron therapy. Nephrol Dial Transplant 14:2680–2687
Safa P, Boulter J, Hales TG (2001) Functional properties of Cav1.3 (alpha1D) L-type Ca2+ channel splice variants expressed by rat brain and neuroendocrine GH3 cells. J Biol Chem 276:38727–38737
LeBeau AP, Robson AB, McKinnon AE, Donald RA, Sneyd J (1997) Generation of action potentials in a mathematical model of corticotrophs. Biophys J 73:1263–1275
Misler S, Barnett DW, Gillis KD, Pressel DM (1992) Electrophysiology of stimulus-secretion coupling in human beta-cells. Diabetes 41:1221–1228
Acknowledgements
We acknowledge the financial support from the Canadian Institute for Health Research (P.H.B. and P.L.). G.Y.O. is a recipient of a Postdoctoral Fellowship from the Canadian Institute for Health Research, Heart and Stroke Foundation of Canada and from the TACTICS program, and P.H.B. is a Career Investigator of the Heart and Stroke Foundation of Ontario.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oudit, G.Y., Trivieri, M.G., Khaper, N. et al. Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J Mol Med 84, 349–364 (2006). https://doi.org/10.1007/s00109-005-0029-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-005-0029-x