Article Text

Abstract
Introduction The National Early Warning Score is used as standard clinical practice in the UK as a track and trigger system to monitor hospitalised patients. Currently, nurses are tasked to take routine vital signs measurements and manually record these on a clinical chart. Wearable devices could provide an easier, reliable, more convenient and cost-effective method of monitoring. Our aim is to evaluate the clinical validity of Polso (ChroniSense Medical, Yokneam Illit, Israel), a wrist-based device, to provide National Early Warning Scores.
Methods and analysis We will compare Polso National Early Warning Score measurements to the currently used manual measurements in a UK Teaching District General Hospital. Patients aged 18 years or above who require recordings of observations of vital signs at least every 6 hours will be enrolled after consenting. The sample size for the study was calculated to be 300 participants based on the assumption that the final dataset will include four pairs of measurements per-patient and per-vital sign, resulting in a total of 1200 pairs of data points per vital sign. The primary outcome is the agreement on the individual parameter scores and values of the National Early Warning Score: (1) respiratory rate, (2) oxygen saturation, (3) body temperature, (4) systolic blood pressure and (5) heart rate. Secondary outcomes are the agreement on the aggregate National Early Warning Score. The incidence of adverse events will be recorded. The measurements by the device will not be used for the clinical decision-making in this study.
Ethics and dissemination We obtained ethical approval, reference number 18/LO/0123 from London—Hampstead Research Ethics Committee, through the Integrated Research Application System, (reference number: 235 034. The study received no objection from the Medicine and Health Regulatory Authority, reference number: CI/20018/005 and has National Institute for Health Research portfolio adoption status CPMS number: 32 532.
Trial registration number NCT03448861; Pre-results.
- health apps
- Mobile Applications [MeSH]
- digital health
- early warning signs
- medical device
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first clinical validation study of a wearable device that measures all National Early Warning Score (NEWS) parameters in a hospital setting.
The study will provide a sufficient number of participants to detect a difference in the primary outcome.
Frequency of measuring NEWS (determined by the NEWS score itself) will not be evaluated, because divergence of scores would complicate clinical care.
Since this validation study uses approved healthcare equipment and nurse lead measurements against a semiautomated wrist-based wearable device with their inherent limitation a careful nurse training and data monitoring/modelling will be required.
Introduction
There is a chronic lack of intensive care unit (ICU) professionals and beds around the world; a recent UK survey found that 4/5 of units had to transfer patients due to lack of beds and 2/5 of units have to close beds due to staffing shortages on at least a weekly basis.1 Delayed admission to ICU for critically ill patients is associated with the increased mortality.2 Therefore, monitoring patients, identifying those who are critically ill and referring them to appropriate units, is of high importance.
Early Warning Scores (EWSs) can be used to determine clinical deterioration of hospitalised patients3 and have shown to perform well in the prediction of cardiac arrest and death within 48 hours.4 EWSs summarise several physiological readings, signs and symptoms into a single parameter, which can then be used to determine specific treatments or refer patients to appropriate care units. This simplifies healthcare practitioners’ assessment of patient status as it helps them to process and evaluate several vital signs.
EWSs are typically used in patients who present to emergency services, and for monitoring thereafter, particularly in acute care units. There are several different versions of the EWS currently under evaluation and in use. The National Early Warning Score (NEWS), developed with the UK’s Royal College of Physicians (RCP), is used as a standard clinical practice in the UK (figure 1).5 The physiological signs of the NEWS include: (1) respiratory rate, (2) pulse rate, (3) oxygen saturation, (4) body temperature, (5) systolic blood pressure, (6) heart rate and (7) level of consciousness.

National Early Warning System table: A, alert; BP, blood pressure; NEWS, National Early Warning Score; NHS, National Health Service; P, pain; U, Unresponsive; V, Verbal.5
Traditionally, NEWS measurements are undertaken by a nurse using separate equipment for different parameters and recording these on a paper chart. Recent efforts have been done on electronically recording these measurements instead of on paper.6–8 Also, continuous monitoring systems have been used in countries such as the USA for certain vital signs.9 These continuous systems algorithmically describe patient status, to monitor and alert when status change thresholds are reached. However, in other countries, including the UK, these measurements are currently typically done manually at specified time points, which takes considerable nursing time and can be inconvenient for patients.
Wearable wrist-based devices can be used to obtain and processes these physiological parameters. Wearable devices could provide an easier, reliable, more convenient and cost-effective method of monitoring patients.6–8 In cases where manual testing is preferred for any parameter, such as the use of supplemental oxygen or level of consciousness, the individual score can be entered into the system manually. There are a number of wrist-based devices able to monitor some of the components of the EWS on the market. However, very few data is available on the use of wearable devices for measurements or monitoring all the components of the EWS in hospital settings.9 10 This study fills this gap by evaluating the clinical validity of Polso, a wrist-based device, as data acquisition and processing platform capable of providing NEWS summaries. The aim of this study is to assess the clinical validity of the Polso wearable device by comparing it to the currently used manual NEWS system.
A validation study will be conducted that consists of two phases: (1) data acquisition for algorithm optimisation; (2) validation (figure 2). The data acquisition for algorithm optimisation calibration phase will provide a learning data set. Physiological readings of Polso will be compared against those taken conventionally and determine whether any data acquisition software algorithms require calibration to achieve an optimal statistical fit for the first 150 patients. If such data acquisition for algorithm optimisation calibration significantly improves Polso performance, ChroniSense will provide a new software package designed for analysis of the second phase. In the second phase, the Polso will be compared with UK-NEWS aggregate score and individual parameter scores. The study conducted parallel with current clinical practice will not use Polso measurements to influence clinical decision-making. This study has been designed to minimise sources of bias by being a blinded study to the clinical staff in charge of the patients’ care. The research team will not be directly involved in the patients’ clinical care.

Adapted consort 2010 flow diagram. A, alert; P, pain; U, unresponsive; V, verbal
The nurse lead measurements on approved healthcare equipment will be delivered by a specially trained ward nurse team paying special methodological attention to the correct acquisition of vital signs. The quality of data will be monitored by the research staff.
The following standard measurements versus Polso measurements will be taken: (1) respiratory rate score (0–3) and value; (2) oxygen saturation score (0–3) and value; temperature score (0–3) and value; systolic blood pressure score (0–3) and value; pulse rate score (0–3) and value; overall score (0–20) (which includes supplemental oxygen yes (2) or no (0)) and level of consciousness alert (A):0, voice (V), pain (P), unresponsive (U): 3.
Scores of 0 are within accepted normal values for each parameter, with some exceptions currently based on clinical presentation. Scores are determined on a per-parameter and aggregate basis. Aggregate scores 4 and below indicate low clinical risk; scores from 5 to 6 indicate medium risk and scores of 7 or higher indicate high risk. In addition, any individual parameter ‘red’ scores (scores with a level of 3) is of at least medium risk, requiring urgent clinical evaluation.
Frequency of clinical monitoring is also determined by the aggregate score, with more frequent monitoring based on higher scores. The current schedules call for reassessment at least every 12 hours for those patients with lower scores, at least every 4–6 hours for those with scores of 1–4, at least every hour for those with scores of 5 or 6 or a score of 3 for a single parameter, and constant monitoring for patients with a score of 7 or higher. Note that these are minimum recommendations, and medical staff can decide on a higher frequency if warranted.
Study participants will be eligible patients admitted to Milton Keynes University Hospital (MKUH) Cardiac Care Unit (CCU). We aim to recruit 300 participants over a maximum of 4-month period; 150 in phase 1 and 150 participants in phase 2 (with a conservative estimate of 1200 measurements to compare). Sample size considerations are based on both power analyses of the main score comparisons and simulation models of the two-phase algorithm optimisation strategy, as detailed in the sample size calculation section. Half of the patients will be in each phase for the most efficient utilisation of patient cohorts.
Methods and analysis
This protocol is reported according to the Standard Protocol Items: Recommendations for Interventional Trials Checklist.11
Study setting
This study will take place at the MKUH CCU. Eligible patients who arrive at MKUH Cardiology Ward (a unit with 26 beds) will be consecutively enrolled after consenting to the study. The CCU also admits patients who are not candidates for ICU but require more than standard ward level frequency of observations and potentially ionotropic support (drugs that affect the strength of contraction of the heart muscle) or non-invasive ventilation (airway support administered through a face mask). Patient length of stay occurs in a bimodal distribution of 2–4 days and 4–6 weeks.
Eligibility criteria
Inclusion criteria will be: a participant who will be willing and able to give informed consent for participation in the study, aged 18 years or above, require recordings of observations of vital signs at least every 6 hours. The patient may not enter the study if any of the following absolute exclusion criteria apply: (1) unable or unwilling to give valid consent for participation in the study; (2) known history of allergy to the strap material (polyurethane); (3) known pregnancy); (4) known essential tremor or Parkinson’s disease; (5) patients with infectious diseases requiring isolation such as methicillin-resistant Staphylococcus Aureus, Clostridium difficile and Extended-Spectrum Beta-Lactamases or (6) any additional condition(s) that in the investigator’s opinion would warrant exclusion from the study or prevent the subject from completing the study. Relative exclusion criteria (exclusion/inclusion of any such patients will be dealt with on a case-by-case basis, at the discretion of the treating physician) are patients deemed ‘clinically unstable’ due to cardiac arrhythmias and/or haemodynamic compromise or patients with irritated skin, injured tissue or open wound over the left wrist.
Data collection
The ChroniSense Polso (figure 3) monitors vital signs, allowing healthcare professionals to manage chronic disease without interfering with the patient’s everyday activity. The POLSO is a wearable wrist device, which uses photoplethysmogram (PPG) and ECG systems, accelerometer and temperature sensors to monitor pulse rate, oxygen saturation, ECG, blood pressure, respiratory rate and body core temperature, plus additional components for the communication.

The POLSO system components.
Polso acquires, derives and processes five physiological parameters and use of supplemental oxygen as well as the level of consciousness can be entered into the system manually (table 1). The Polso wrist watch-like device will be sized and placed as comfortably as possible on the patients' wrist. According to the NEW/RCP recommendations, patients typically have measurements of vital signs taken at least every 6 hours in those who are deemed clinically stable, with the frequency of up to every 5 min in those patients who are clinically deteriorating. When the patient is due to have conventional vital signs measured, Polso measurements will be recorded at the same time. The measurements will be coordinated and synchronised between the specially trained ward nurses and the research team who will be responsible only for their respective data measurements either by the commercial healthcare equipment or with the wrist wearable device, respectively. To overcome the intrapatient variability of blood pressure and heart rate readings during traditional (Riva-Rocchi) blood pressure measurements—readings with the Polso device will be taken from the same arm for all variables immediately before and after the traditional blood pressure measurements. Participants will be in the study for up to 72 hours after recruitment into the study.
Summary of how Polso processes each National Early Warning Score (NEWS) parameter
Frequency of clinical monitoring is also determined by the aggregate score, with more frequent monitoring based on higher scores. The current schedules call for reassessment at least every 12 hours for those patients with lower scores, at least every 4–6 hours for those with scores of 1–4, at least every hour for those with scores of 5 or 6 or a score of 3 for a single parameter, and constant monitoring for patients with a score of 7 or higher. These, of course, are minimum recommendations, and medical staff can decide on a higher frequency if warranted. Frequency of measuring NEWS (determined by the NEWS score itself) will not be evaluated, because divergence of scores would complicate the clinical evaluation. To comply with the Good Clinical Practice for ongoing patient care the currently used NEWS scores will be taken into account and not the experimental Polso derived observations.
Outcomes
Primary outcomes will be statistical agreement for the on the individual parameter scores (as assessed by kappa above criterion) and values (as assessed by Bland-Altman limits of agreement within criterion): (1) respiratory rate, (2) oxygen saturation, (3) body temperature, (4) systolic blood pressure and (5) pulse rate (table 2).
Acceptable accuracy of Polso parameters
There are no accuracy standards for pulse and respiration, therefore, the accuracy of suitable reference devices is specified.
Secondary outcomes will be: (1) performance: to evaluate the performance of Polso as a data acquisition and processing platform capable of providing aggregate EWS score, against the currently employed NEWS system (as based on kappa statistical criteria as outlined below); (2) performance: to use the data obtained from phase 1 to calibrate the modelling of any deficient parameter so that its accuracy is significantly improved. Ideally, this represents the performance within acceptable limits as defined by regulatory standards or performance of comparable measurement systems. Safety outcomes will be the incidence of adverse events (AEs), as defined by the description below. Data of the patients collected until the AE will be used unless the patient has revoked their consent.
Adverse device event
AE related to the use of an investigational medical device. This includes any AE resulting from insufficiencies or inadequacies in the instructions for use, the deployment, the implantation, the installation, the operation or any malfunction of the investigational medical device or any event that is a result of a use error or intentional misuse.
Adverse event
Any untoward medical occurrence, unintended disease or injury or any untoward clinical signs (including an abnormal laboratory finding) in subjects, users or other persons whether related to the investigational medical device. This includes events related to the investigational device or the comparator and events related to the procedures involved (any procedure in the clinical investigation plan). For users or other persons, this is restricted to events related to the investigational medical device.
All AEs will be graded for severity as followed: (1) mild—events require minimal or no treatment and do not interfere with the participant’s daily activities; (2) moderate—events result in a low level of inconvenience or concern with the therapeutic measures. Moderate events may cause some interference with functioning; (3) severe—events interrupt a participant’s usual daily activity and may require systemic drug therapy or other treatment. Severe events are usually potentially life-threatening or incapacitating.
Definitions of serious AEs
Serious AE (SAE) AE that: (1) led to a death; (2) led to a serious deterioration in health that either resulted in a life-threatening illness or injury, or resulted in a permanent impairment of a body structure or a body function, or required in-patient hospitalisation or prolongation of existing hospitalisation, or resulted in medical or surgical intervention to prevent life-threatening illness or injury or permanent impairment to a body structure or a body function; (3) led to fetal distress, fetal death or a congenital abnormality or birth defect. This includes device deficiencies that might have led to an SAE if: (1) suitable action had not been taken or; (2) intervention had not been made or; (3) if circumstances had been less fortunate. These are handled under the SAE reporting system.
Definition of unanticipated serious adverse device effect
Unanticipated serious adverse device effect serious adverse device effect which by its nature, incidence, severity or outcome has not been identified in the current version of the risk analysis report.
Time period and frequency for event assessment and follow-up
The occurrence of an AE or SAE may come to the attention of study personnel during study admission of a study participant presenting for medical care or on review by a study monitor. All AEs including local and systemic reactions not meeting the criteria for SAEs will be captured on the appropriate case report form (CRF). Information to be collected includes event description, time of onset, clinician’s assessment of severity, relationship to study product (assessed only by those with the training and authority to make a diagnosis) and time of resolution/stabilisation of the event. All AEs occurring while on the study must be documented appropriately recorded on the AEs page of the CRF. Severity and relationship to study device will be assigned by the investigator regardless of relationship. All AEs will be followed to an adequate resolution.
Changes in the severity of an AE will be documented to allow an assessment of the duration of the event at each level of severity to be performed. AEs characterised as intermittently require documentation of onset and duration of each episode. The principle investigator will record all reportable events with start dates occurring any time after informed consent is obtained and throughout the study including the follow-up termination visit.
Reporting SAE s
Any SAE, whether deemed device-related or not, must be reported to the site ethics committee and to the sponsor’s Vice President regulatory by telephone and by email, as soon as possible after the investigator has become aware of its occurrence even if not all the information is available at the time of initial contact. The investigator must complete an SAE Form, and send it, via email, to the sponsor within 24 hours of becoming aware of the event. Accompanying documentation, such as copies of hospital case reports, autopsy report and other documents when applicable should be sent as soon as they are available. The site’s ethics committee must also be duly notified and dealt with, according to the Hospital and Medicine and Health Regulatory Authority regulations. Subjects who have had an SAE must be followed clinically until all parameters (including laboratory) have either returned to normal or are stabilised.
Sample size calculation
It is estimated that a sample of 300 participants is sufficient for the aims of this study. If the final dataset includes four pairs of measurements per-patient and per-vital sign, this results in a total of 1200 pairs of data points per vital sign. Agreement between the two methods for each vital sign will be assessed through an appropriately modified version of the Bland-Altman methodology taking into account intrasubject repeated measurements.12 With 300 patients and intrasubject variance larger than 10% of the total variance, this methodology can detect agreement between the two methods with 95% CI. In the extreme case where the intrasubject measurements are perfectly correlated (ie, the intrasubject variance is 0), the study remains well powered. Alternatively, a paired t-test can reveal small differences (ie, a bias equal to 20% of the total variance) between the two methods with 93% power. This assumes the extreme case, where the intrasubject measurements are perfectly correlated and, therefore, the effective number of data points is 300. In practice, the effective sample size is larger than 300 and, therefore, the power is expected to be even higher.
In addition to the above approach, we shall also use the kappa statistic to assess agreement along the ordinal scale of NEWS outcomes.13–15 Assuming that kappa ≥0.90 indicates agreement, while kappa ≤0.75 indicates disagreement, our analysis has power at least 80% at a Bonferroni-adjusted significance level (for five parameters) equal to 0.01. This is indicated in table 3, where we show the number of data pairs required for achieving power 80% for 3, 4 or 5 NEWS categories and for both equal or unequal distribution of measurements across these categories. The maximum number of required measurements is 219.
Required number of paired measurements to achieve power 80% using the kappa statistic
Four of the five parameters required for computation of the NEWS are already at specified performance in preliminary UK National Measurement Laboratory and company bench studies. The remaining parameter, systolic blood pressure, is nearly at performance level, but still requires calibration tuning, which in turn requires additional data. It is proposed to use all available data from the first phase of the study in the calibration of our models, as data become available. The overall cohort of 300 patients, called for by the limit of agreement analysis, will be split into two equal halves: one for data acquisition for algorithm enhancement, the other for validation. This agrees with common practice for optimisation of data acquisition for the algorithm enhancement/validation modelling procedures. Statistical bootstrap simulation verified this optimisation against healthy subject data. Figures are provided illustrating the results of these simulations performed on both systolic and diastolic blood pressure data, both indicating that the optimum proportion of data to assign to the validation stage is 50%.
The curve in figures 4 and 5 shows the effect of varying the percentage overall data used in the validation phase (x-axis) on the error of model calibration (mean absolute error, y-axis). The optimum division of the overall cohort, where the error is minimised, is at 50%.

Results of systolic pressure bootstrap simulation studies on healthy subject data (indicating optimal calibration/validation cohort breakdown).
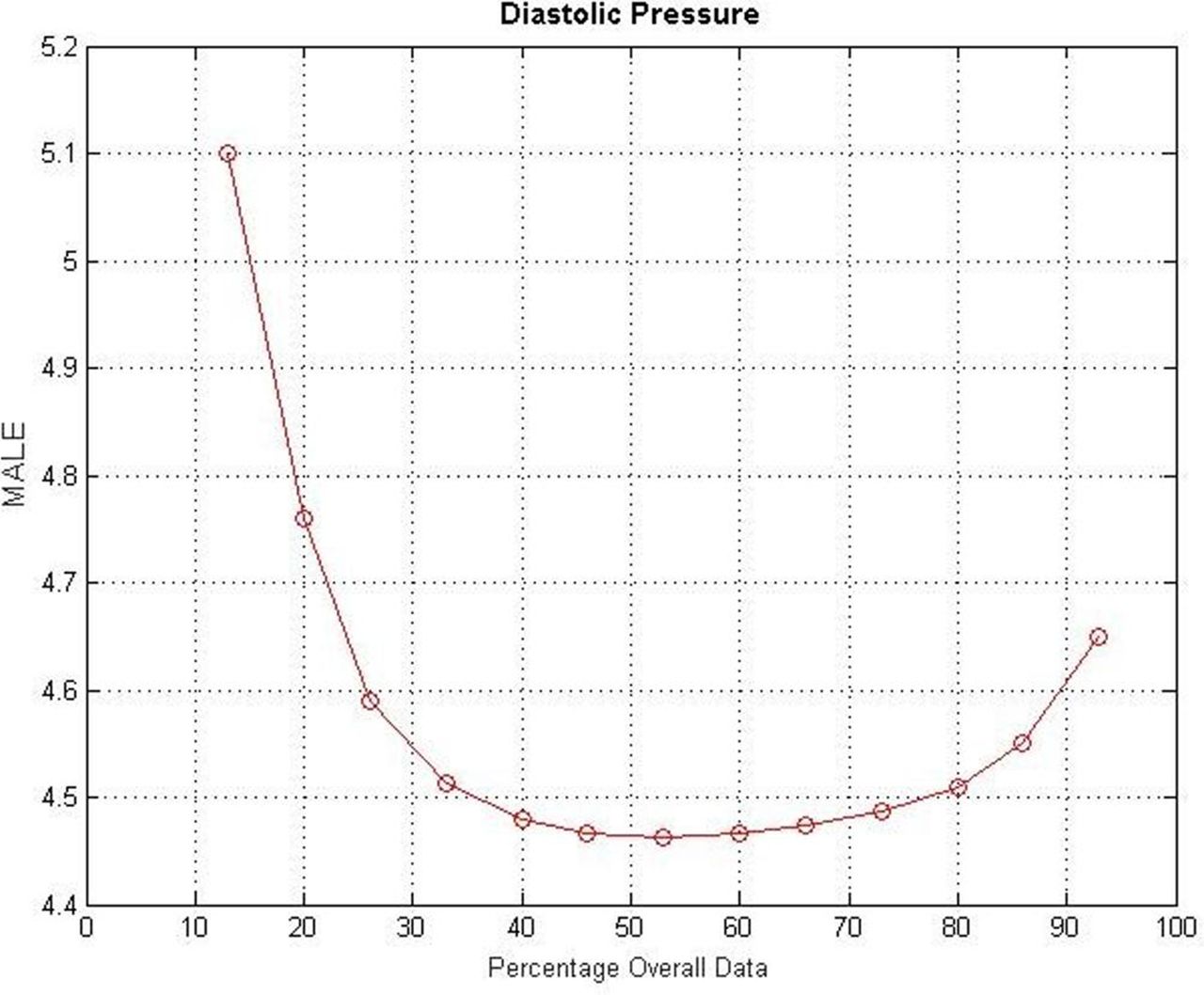
Results of diastolic pressure bootstrap simulation studies on healthy subject data (indicating optimal calibration/validation cohort breakdown). MAE; mean absolute error.
Study schedule
The additional processes that will occur in addition to the standard care will be as follows (figure 6). Patients will be identified by the cardiology research fellow and research nurse who are part of the clinical cardiology team. If patients are transferred to ICU or other wards within MUKH, their data will be used but we will not be able to continue to monitor them. Participants will be approached and given information regarding the study on the day of arrival (admission). They will have the opportunity to ask further questions and if they agree to take part will be asked to complete a consent form (online supplementary file). Written informed consent will be obtained and the patient will be allocated a study number. Relevant participant demographic and baseline characteristics will be recorded in the CRF. Information that will be collected at baseline is the reason for admission MKUH CCU or ward 17, patient’s anthropometrics and demographics (age, gender, height and weight), and previous medical history. The researcher will place the device on the wrist of the patient after informed consent has been given. The researcher with will perform size adjustment of the wrist band to allow for the maximum comfortability of the wearer. The researcher will record standard parameters of blood pressure, pulse rate, respiratory rate, oxygen saturations and body temperatures obtained by the Polso. The EWS will be calculated from these measurements. The study participation will not alter the normal clinical progress of the subject through hospitalisation. Recordings of vital signs will be extracted from the Polso while standard recordings are obtained by the nursing team. Patients will be wearing the device on their wrist and there will be no interference with the process of obtaining standard measurements. Each participant has the right to withdraw from the study at any time. In addition, the investigator may discontinue a participant from the study at any time if the investigator considers it necessary for any reason including: ineligibility (either arising during the study or retrospectively if not known at the time of screening), withdrawal or loss of consent, lost to follow-up. The reason for withdrawal will be recorded in the CRF. End of the study will be defined as the date and time when the final reading of the last subject was done.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The flow of participants through the study.
Data monitoring methods
Source data will be collected using electronic CRFs (e-CRFs) for each participant by the research team and the anonymised data entered onto a secure electronic database. The database and e-CRFs on secured network computers at the hospital will be accessed by the research team only.
Data monitoring
The formation of an independent data monitoring committee has been deemed unnecessary for this low risk, non-interventional study to be conducted over a short time frame. However, a group comprising of a statistician, research fellow and sponsor representative will review data management processes during the study. The group will advise the trial management group as to whether there is evidence or reason why the study should be amended or terminated based on recruitment rates or data inconsistencies or safety. Interim analyses will be performed by the study team and results made available to Trial Management Group to make any final decisions to terminate the study.
Harms
Any AEs and/or unintended effects arising from conducting the study will be reported as per the study AE reporting framework. Device-related adverse and/or unintended effects arising from conducting the study will be reported in accordance with device risk and safety reporting plan.
Auditing
The sponsor (MKUH) will carry out data monitoring and auditing in accordance with protocol and data monitoring plan. Planned auditing will be carried out at least twice during the study.
Statistical methods
Descriptives
We will descriptively analyse characteristics of participants including the reason for admission MKUH CCU, patients’ anthropometrics and demographics (age, gender, height and weight) and medical history. Baseline characteristics of the participants will be reported for: (1) all participants enrolled and (2) participants included in the analysis. Categorical variables will be described using frequencies and proportions. Continuous variables will be described using means and SD if the distribution of the variable is normally distributed (observed or transformed). If data are not normally distributed, medians and the lower and upper quartile will be reported.
Outcome analysis
Individual NEWS parameters will be evaluated by limits of agreement (Bland-Altman analysis). Values of agreement and 95% CI will be reported. Performance within the limits of agreement specified by regulatory standards or performance achieved by comparable devices will be used as the criterion for individual parameters. Such limits are provided in table 2.
Kappa agreement analysis will be carried out for aggregated NEWS scores. Kappa of 0.90 or greater will be used as the criterion for substantial agreement between Polso-based and standard NEWS methods. The agreement is the degree to which scores are identical. Cohen’s kappa and derivative statistics can be used to indicate the strength of agreement for nominal and ordinal scales used on separate occasions. Cohen’s kappa compares the observed agreement with an agreement that is expected by chance alone, which makes it a chance-corrected index of agreement. A kappa value of 0 means that there is no agreement beyond chance, while a kappa value of 1 indicates that there is perfect agreement. There is no accepted standard for rating the different values for kappa. Kappa values higher than 0.60, 0.70 or 0.80 are generally considered to be the minimum standard for group-level comparisons or for research purposes. However, these strengths of agreement do not indicate the practical relevance of results. Recommendation for quantitative interpretation of kappa applies to the social rather than medical sciences. For example, Fleiss (1) indicated values over 0.75 as ‘excellent’, but this is not justified on any realistic statistical grounds for the medical sciences where criteria need to be stricter. We propose two considerations in determining our relevant criterion levels of kappa. First, the NEWS relies on nominal or ordinal scales which are transformations from quantitative measures. Disagreements will only be relevant at boundary values between individual NEWS subscore ratings; for quantitative scores which are already in near-agreement, these should constitute only a small proportion of data. Second, the medical consequence of disagreement; more frequent monitoring, a consequence of worsening NEWS scores, is not so problematic, but failure to detect deterioration may have severe consequences. Both considerations suggest that an extremely high value of kappa be considered, on the order of 0.90 or greater. Such high values of kappa (indicating a strong statistical effect) are associated with smaller numbers of observations to achieve statistical significance than are considered for the other measures in this study. Therefore, these were not addressed directly in the determination of sample size.
There are additional considerations for the ordinal data. Specifically, disagreements between different ratings are not equally important for ordinal data. To take this into account, Cohen introduced weights for the calculation of weighted kappa. Weighted kappa takes account of the distance between disagreements and is therefore appropriate for ordinal scales with more than two categories. Different weights can be given to weighted kappa, but most commonly used are Cicchetti-Allison and Fleiss-Cohen weights. Fleiss-Cohen gives quadrant weights and can be similar to the intraclass correlation coefficient. Cicchetti-Allison gives linear weights and is more appropriate for questions with many answer options. The linearly weighted kappa coefficient can be simply derived from K-1 embedded 2×2 classification tables.
NEWS parameters will be analysed by weighted kappa statistics (treating the scores as ordered categories), using Cicchetti-Allison methods. Analyses will primarily be performed on a per-patient basis, with results pooled over patients. Appropriate weighting methods, due to different numbers of readings and different durations of hospitalisations per patient, and their relations to severity must be employed. Analyses will have to be performed twice should ChroniSense determine that significant improvement is possible based on the training set cohort, where significant improvement refers to bringing accuracy in line with acceptable limits. In that case, the test set cohort analysis will provide definitive results. Similarly, evaluation of how differences in NEWS between Polso and UK-NEWS might affect patient care will require careful statistical design and planning.
Handling missing data
All available data will be included in the analysis of the outcomes. The frequency and percentage of lost and/or failed measurements will be reported.
Handling outliers
Residual outliers will be identified using assumption checks. If any residual outliers are identified, a sensitivity analysis will be conducted where the analysis is repeated with the outliers excluded from the analysis. If there is no difference in conclusions, the analysis with the outliers included will be reported otherwise both analyses will be reported.
Patient and public involvement
Not undertaken for this part of the study.
Ethics and dissemination
ChroniSense Medical notified the Medicines and Health Regulatory Authority of this study (MHRA reference number CI/20018/005). The results of this study are planned to be used for medical device approval in Europe (CE-marking). The Polso is pre-CE mark and as such is for clinical investigation use only. This will be clearly labelled on the system and in the associated Clinical User Manual. The system will have a part number, S/N/LOT number for identification, in accordance with the requirements of the standard ‘EN 1041:2008’ information supplied by the manufacturer of medical devices and ISO 14155:2011, para 5.10 and UK regulations for labelling of the investigational devices. The location of labelling will be clearly indicated in the product’s Instruction for Use. Product labelling will include; the name and address of the manufacturer and all other essential information to allow full traceability of the product.
Protocol amendments
Deviations from the protocol will be discussed with the sponsor and described in the final study paper.
Informed consent
A member of the research team (research fellow or research nurse) will approach eligible patients and give a brief explanation of the study, to assess interest in the study. The patient information sheet will be given if the participant agrees to participate in the study. Once the participant has had time to read the information, the research team member to return to discuss the study offering the participant the opportunity to ask questions before deciding whether to participate. Agreement to participate will be recorded using the study consent form, which will be signed and dated by the participant and research team member.
The consent form will be kept by the investigator and a copy will be given to the participant to keep. We will not be approaching adults who are unable to give consent or are part of vulnerable groups. The participant must personally sign and date the latest approved version of the informed consent form before any study-specific procedures are performed.
Written and verbal versions of the participant information and informed consent will be presented to the participants detailing: the exact nature of the study; what it will involve for the participant; the implications and constraints of the protocol; the known adverse effects and any risks involved in taking part. It will be clearly stated that the participant is free to withdraw from the study at any time for any reason without prejudice to future care, and with no obligation to give the reason for withdrawal. The participant will be allowed enough time to consider the information, and the opportunity to question the investigator, their general practitioner or other independent parties to decide whether they will participate in the study.
Written informed consent will then be obtained by means of participant dated signature and dated signature of the person who presented and obtained the informed consent. The researchers who obtained the consent will be suitably qualified and experienced and have been authorised to do so by the chief investigator. A copy of the signed informed consent will be given to the participant. The original signed form will be retained at the study site.
Confidentiality and access to data
Patient-identifiable information will only be accessible to members of the research team. Paper documents containing patient identifiable information such as consent forms, patient identification logs and source data will be filed in a secure location at the hospital accessible to members of the research team only. All data will be anonymised prior to analysis by allocating a trial number to each participant.
Participant safety
There is no anticipated risk to patients or researchers from taking part in this study. A formal risk analysis assessment has been conducted for the Polso in accordance with the standard EN ISO 14971:2012 Medical Devices. Application of risk management to medical devices’ and a risk management file has been generated. No adverse side effects from the use of the device have been identified.
The Polso meets all relevant safety-related essential requirements as defined by the Medical Device Directive 93/42/EEC, Essential Requirements Checklist. The safety of the Polso has been identified and demonstrated during the risk assessment, design verification testing and compliance with applicable standards.
The Polso has been subjected to electrical safety testing and several feasibility studies, to demonstrate its safety and performance. The combined results indicate that the Polso is safe. All components of the Polso are designed, manufactured and tested according to relevant international standards for medical devices. The device has been calibrated by the National Physical Laboratory (the UK's National Measurement Institute and a world-leading centre of excellence in developing and applying the most accurate measurement standards, science and technology) with healthy subjects for ECG, oxygen saturation, pulse rate and blood pressure-related parameter.
Dissemination policy
The study is planned to be published in a peer-reviewed Journal, presented at conferences and also published on websites (the MKUH National Health Service Foundation Trust, Oxford University and ChroniSense Medical).
Footnotes
MHVV and FA are joint first authors.
GW, DB and AK are joint senior authors.
Contributors MHVV wrote the protocol, FA revised the protocol, DV conducted the power calculation, GW and DB commented on the protocol. The concept of the study was conceived as well as the protocol revised and corrected by AK.
Funding This study was supported by the SENS Research Foundation with a donation from ChroniSense Medical.
Disclaimer The funder did not influence the study design; and will not influence data collection, management, analysis and interpretation of data; writing of the report; and the decision to submit the report for publication.
Competing interests The research sponsor for this study is Milton Keynes University Hospital (MKUH) NHS Foundation Trust and collaborator is Oxford University NHS Foundation Trust. DB is a stockholder in Translation Ventures. (Charlbury, Oxfordshire, UK) and IP Asset Ventures (Oxford, Oxfordshire, UK), companies that, among other services, provide cell therapy biomanufacturing, regulatory and financial advice to pharmaceutical clients. DB is also subject to the CFA Institute’s codes, standards and guidelines, so, he must stress that this piece is provided for academic interest only and must not be construed in any way as an investment recommendation. Additionally, at the time of publication, DB and the organisations with which he is affiliated may or may not have agreed and/or have pending funding commitments from the organisations named here.
Patient consent for publication Not required.
Ethics approval We obtained ethics approval through the Integrated Research Application System (IRAS) for the CHESS: ChroniSense National Early Warning Score Study, reference number: 18/LO/0123.
Provenance and peer review Not commissioned; externally peer reviewed.