Article Text

Abstract
Objectives There is sparse information on the safety of early primary discharge from the emergency department (ED) after rule-out of myocardial infarction in suspected acute coronary syndrome (ACS). This prospective registry aimed to confirm randomised study results in patients at low-to-intermediate risk, with a broader spectrum of symptoms, across different institutional standards and with a range of local troponin assays including high-sensitivity cTn (hs-cTn), cardiac troponin (cTn) and point-of-care troponin (POC Tn).
Design Prospective, multicentre European registry.
Setting 18 emergency departments in nine European countries (Germany, Austria, Switzerland, France, Spain, UK, Turkey, Lithuania and Hungary)
Participants The final study cohort consisted of 2294 patients (57.2% males, median age 57 years) with suspected ACS.
Interventions Using the new dual markers strategy, 1477 patients were eligible for direct discharge, which was realised in 974 (42.5%) of patients.
Main outcome measures The primary endpoint was all-cause mortality at 30 days.
Results Compared with conventional workup after dual marker measurement, the median length of ED stay was 60 min shorter (228 min, 95% CI: 219 to 239 min vs 288 min, 95% CI: 279 to 300 min) in the primary dual marker strategy (DMS) discharge group. All-cause mortality was 0.1% (95% CI: 0% to 0.6%) in the primary DMS discharge group versus 1.1% (95% CI: 0.6% to 1.8%) in the conventional workup group after dual marker measurement. Conventional workup instead of discharge despite negative DMS biomarkers was observed in 503 patients (21.9%) and associated with higher prevalence of ACS (17.1% vs 0.9%, p<0.001), cardiac diagnoses (55.2% vs 23.5%, p<0.001) and risk factors (p<0.01), but with a similar all-cause mortality of 0.2% (95% CI: 0% to 1.1%) versus primary DMS discharge (p=0.64).
Conclusions Copeptin on top of cardiac troponin supports safe discharge in patients with chest pain or other symptoms suggestive of ACS under routine conditions with the use of a broad spectrum of local standard POC, conventional and high-sensitivity troponin assays.
Trial registration number NCT02490969.
- registry
- acute coronary syndrome
- myocardial infarction
- copeptin
- troponin
- mortality
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first large European registry demonstrating the safety of the dual marker strategy using cardiac troponin and copeptin for early discharge in patients with suspected acute coronary syndrome.
The study recruited less selected patients, a broader range of local cardiac troponin (cTn) assays and assay generations and across different institutional standards than former studies and thus reflects daily routine in clinical practice.
The study has been carried out in experienced centres, thus in settings with lower clinical expertise results may differ.
The very low mortality rate does not allow any exploratory analyses on the safety of discharge by centre volumes, experience of physicians, local cTn assay or assay generation.
Introduction
Chest pain accounts for approximately eight million annual emergency department (ED) visits in the USA,1 rendering chest pain the second most common presenting symptom. In a pooled analysis on 51 observational trials, the prevalence of the final diagnosis of acute coronary syndrome (ACS) was confirmed in a median of 14%, with a range from 5% to 42%.2
An effective risk stratification is paramount to select the most appropriate decision for admission or direct discharge because admission of patients at low or very low risk is not safe3 4 as it increases the risk to receive unnecessary coronary angiography, coronary interventions, multiple re-admissions3 and eventually the risk of periprocedural myocardial injury or type 4 myocardial infarction (MI), and procedure-related major bleedings.4 Moreover, unselected admission of chest pain patients for further workup for the evaluation of ACS is time consuming and costly.5 6 During an interval of only 9 years (from 1999 to 2008), the use of advanced medical imaging for ED visits related to chest pain was found to increase dramatically by 367.6% in the CDC/NCHS (Centers for Disease Control and Prevention/National Center for Health Statistics), National Hospital Ambulatory Medical Care Survey.7 On the other hand, early discharge is also not without risk, as up to 2% to 5% of patients with ACS are reported to be inappropriately discharged from the ED every year5 8 although the methodology to assess these numbers is limited (no complete follow-up of all patients, no exact differentiation between incident and prevalent acute myocardial infarction (AMI) and the components of ACS). Nevertheless, missed or incident AMI early after discharge is associated with a HR for death of 1.7% to 1.9%.8 Missed AMIs account for 20% of USA emergency medicine related litigation dollars.9 Currently, use of high-sensitivity cardiac troponins (hs-cTn) has improved the accuracy and earlier detection of an MI,10–13 and very low concentrations of hs-cTn have been reported to safely rule-out an MI and to be associated with rates of death or MI below 1%.14–17 Accordingly, 2015 European Society of Cardiology (ESC) guidelines on acute coronary syndrome without ST-segment elevation (NSTE-ACS)10 discourage routine coronary angiography in low risk patients and recommend early discharge after clinical risk stratification, and a pre-discharge or post-discharge stress imaging test for the decision of a selective invasive strategy. Supporting evidence for early uneventful discharge of low risk patients stems mainly from observational studies14 15 18 19 where investigators were commonly blinded to the investigational hs-cTn results, were unaware of retrospectively derived optimal decision cut-offs, and managed patients at their own discretion following standards of care applicable at that time. In fact, most of the patients who retrospectively fulfilled early rule-out criteria were kept in hospital and neither medical measures nor non-cardiac diagnoses are reported. Only few interventional clinical trials evaluated the safety of a randomised allocation to early discharge versus conventional care in patients at low20 21 or low-to-intermediate high risk.22 The Biomarkers-in-Cardiology 8 (BIC-8) trial22 tested the utility of a dual biomarker strategy using normal cTn or hs-cTn values, that is below the upper limit of normal, mainly the 99th percentile, together with normal Copeptin values below the 95th percentile (<10 pmol/L) to identify candidates for direct early discharge from the ED. The findings demonstrated that this strategy reduced the length of observation time in the ED or chest pain unit and increased rates of discharge at a low risk for major adverse cardiovascular events that was comparable or even lower in the per protocol analysis to standard of care. Compared with serial troponin-based protocols, advantages of the dual marker strategy include the ability of instant rule-out of MI without the need for additional blood draw, high sensitivities and negative predictive values (NPVs) for AMI of Copeptin in combination with conventional or contemporary sensitive cTn assays,23–28 or point-of-care troponin (POCT),29 particularly when hs-cTn or validated hs-cTn assays are not available, and supporting data for a safe discharge from a large, appropriately powered randomised multicentre trial.22 The value of Copeptin on top of detectable but still normal cTn or hs-cTn for rule-out of MI has been studied extensively and the dual marker strategy (DMS) algorithm has been quoted as an additional option for instant rule-out in 2015 ESC guidelines.10 In contrast, there is sparse information from randomised trials on the safety of discharge20 21 and the safety of discharge using a prespecified algorithm has rarely been investigated in a prospective registry.
Therefore, the aim of the present multicentre observational trial was to confirm the safety of this strategy that was previously reported in a randomised interventional trial22 in routine clinical practice, across a broad spectrum of cTn assays including POCT, in an unselected population with a broader range of symptoms, and at low-to-intermediate risk presenting with suspected ACS to 18 EDs in Europe and Turkey.
Methods
The Pro-Core is a multicentre, international observational trial with 18 participating centres (online supplementary figure 1S) in Europe and formally Near East (Ankara, Turkey).
Supplemental material
We enrolled adult men and women who present to an ED or chest pain unit (CPU) with signs and symptoms suggestive of NSTE-ACS. Eligible patients qualifying for the DMS strategy were recruited consecutively but entry was restricted to patients with a low or intermediate GRACE (Global Registry of Acute Coronary Events) score.
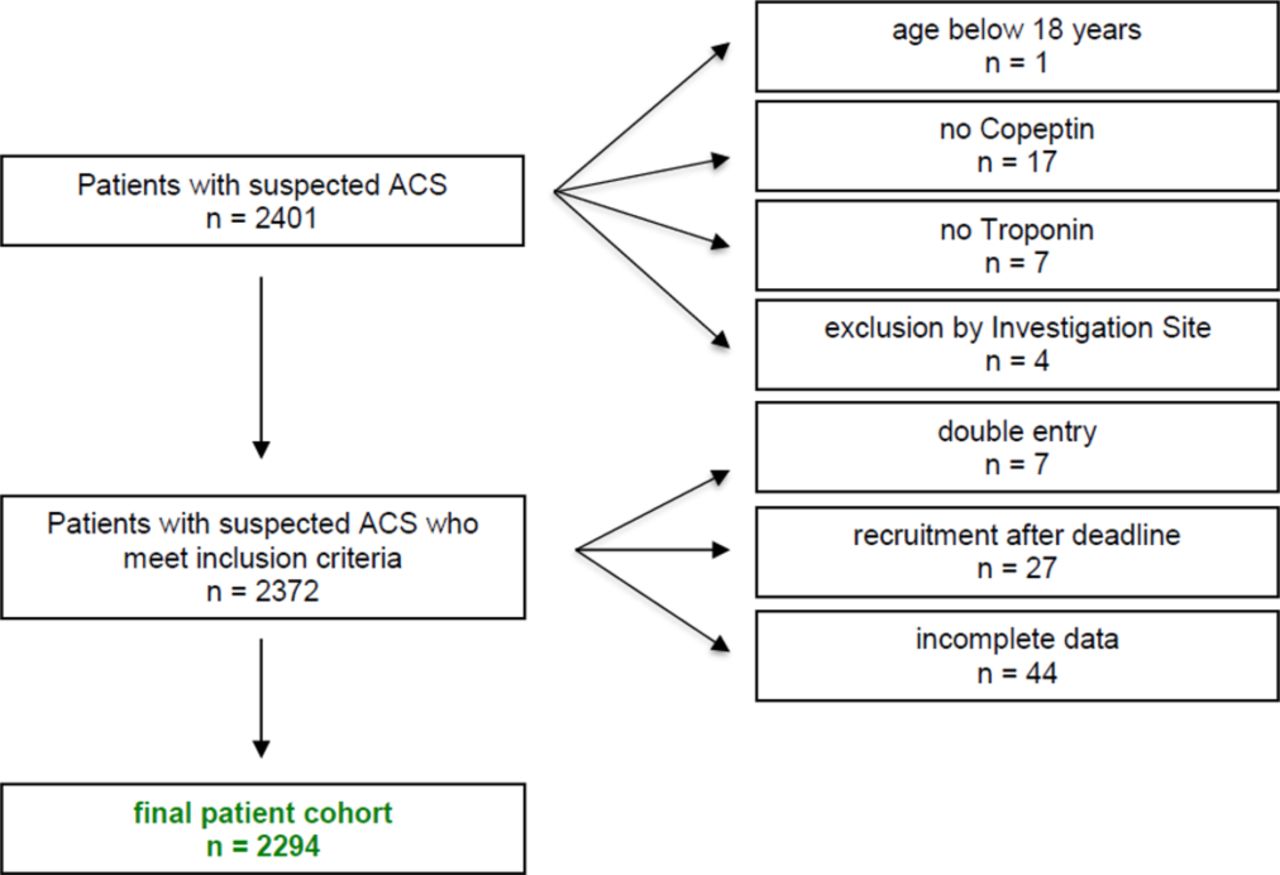
Patients were eligible if they were aged ≥18 years, presented with symptoms suggestive of ACS such as acute chest discomfort, angina pectoris or dyspnoea as leading symptoms. Patients presenting with ST-segment elevation or a final diagnosis of ST-segment elevation myocardial infarction (STEMI) were excluded from analysis (see figure 1 for patient flow).

Patient flow chart. ACS, acute coronary syndrome.
Patients underwent clinical assessment that included medical history, physical examination, standard blood test including measurements of local (hs)-cTn, Copeptin and 12-lead ECG. Baseline information included the Killip class, and clinical information to calculate the GRACE score. Other clinical scores were not tested prospectively prohibiting any conclusion on their clinical usefulness. Physicians had access to all clinical information including Copeptin and cTn results that were reported with local turn-around-times. Decision for primary discharge after rule-out using the dual biomarker strategy, or for disposition of patients if MI was not ruled out was left at the discretion of the attending physician. Patients were excluded if high risk features were evident (eg, the GRACE score was above 140) and if hospital admission was obviously necessary at presentation for any reason. Final diagnosis of NSTE-ACS was performed by the ED physician applying the criteria of the third universal definition of AMI.30 Unstable angina was diagnosed in the presence of new or worsening symptoms of suspected myocardial ischaemia but either normal or undetectable cTn concentrations in serial blood draws, or a cTn together with a Copeptin below the decision limit at presentation. Importantly, classification of ACS was done by the treating physician and was not subject of retrospective adjudication. All patients were contacted at 30 days to assess all-cause mortality. Number of patients was limited to 300 patients per participating site to limit centre bias.
Biomarkers and rule-out algorithms
Copeptin and cardiac troponin were tested from fresh unfrozen blood from a single blood sample drawn at admission to the ED or CPU as part of the routine patient management.
Copeptin was measured using the automated fluoro-immunoassay B∙R∙A∙H∙M∙S Copeptin proAVP KRYPTOR for the quantitative measurement of C-terminal pro-arginine-vasopressin (CT-proAVP, Copeptin) in human serum and plasma on the B∙R∙A∙H∙M∙S KRYPTOR compact PLUS platform. The test has a detection limit of 0.69 pmol/L and a functional assay sensitivity (detected by inter-assay precision of 20% CV) of 1.08 pmol/L.
The recommended cut-off for the decision between a positive and a normal test is 10 pmol/L, corresponding to the 95th percentile of a healthy reference population. This cut-off was used in the randomised controlled trial by Möckel et al,22 and is the recommended cut-off for the rule-out algorithms for MI.
Cardiac troponin was measured at the individual institutions according to standard practice. An overview on local assays and cut-offs is provided as online supplementary table 1S). Briefly, Roche Elecsys hs-cTnT was used in 39%, followed by Abbott Architect hs-cTnI, Siemens (Vista, Loci), Beckman Access TnI, and Radiometer (third generation cTnT) in 22%, 22%, 11% and 6%, respectively. Conventional and high-sensitivity assays were permitted for the early rule-out strategy.
Baseline characteristics of the patients
A patient qualified as rule-out and for early discharge if he presented with signs and symptoms suggestive of ACS, together with a low-to-intermediate risk profile defined as the absence of high risk features (eg, a GRACE score <140), and a combined negative testing of Copeptin and troponin, defined as Copeptin below 10 pmol/L and cardiac troponin below the local AMI decision limit as recommended by the guidelines, mostly the 99th percentile value of a healthy reference population provided by the manufacturer.
Follow-up and clinical endpoints
The primary objective was to evaluate 30 day all-cause mortality in patients in whom acute myocardial infarction was ruled-out using the early dual marker rule-out strategy and who are therefore directly discharged from the ED. All-cause mortality was preferred over cardiovascular death because collection of information is more convenient and because the majority of eligible patients presented to the EDs with non-coronary and non-cardiac diagnoses.
The secondary objectives were evaluated in all patients, irrespective of biomarker test results and disposition. Secondary endpoints included the diagnosis of acute myocardial infarction, final hospital diagnoses, time to discharge/transfer from the ED/CPU, disposition decision (discharge or admission), length of hospital stay, intensive care unit-treatment, performance of coronary angiography/percutaneous coronary intervention (PCI)/coronary artery bypass grafting (CABG), performance of ECGs, stress testing, imaging, performance of cardiovascular monitoring, in-hospital all-cause mortality, 30 day all-cause mortality.
The study protocol also addressed patients where the protocol was violated, that is, those who were not primarily discharged or not admitted although criteria were fulfilled (over-rule). The reasons for over-rule or other protocol violations were registered.
The study complies with the Declaration of Helsinki. The positive vote was sent to all study sites. The principle investigator decided based on local and national rules, whether a separate local ethics committee submission was necessary. Additional ethics approvals were obtained from the sites listed in the online supplementary table 2S. The ethics committee approved that anonymised routine data of patients were used without informed consent for this registry. The study was registered before enrollment of the first patient.
Supplemental material
Statistical evaluation
Enrolment was restricted to a maximum number of 300 patients per centre to ensure generality by avoiding the dominance of single centres. The total number of patients enrolled therefore depended rather on the number of participating centres than on their enrolment performance. As the primary objective of this registry was the monitoring of an already routinely applied clinical algorithm, no confirmatory study design was chosen and there was no sample size calculation performed. An exploratory analysis of the safety of DMS by local cTn assay or assay generation, or by study centre was not done as there was only one death precluding meaningful analysis. All data were entered into an online electronic case report form. Group comparisons for categorical variables were performed using X2 tests and for numerical variables using Wilcoxon rank-sum tests. A p value below 0.05 was considered significant (no correction for multiple testing conducted) and 95% CIs were determined for binary all-cause death at 30 days by the method of Clopper and Pearson and for numeric length of stay in the ED/CPU by 2.5%-quantiles and 97.5%-quantiles estimated by bootstrapping.
Statistical analyses were performed using the software R V.3.1.2 and SPSS (IBM SPSS Statistics, V.21).
Patient and public involvement
Patients or public were not involved in the development of the study protocol.
Results
A total of 2401 consecutive patients with suspected ACS were screened from 16 September 2015 until the end of recruitment on 23 May 2017. Of these, 107 patients were excluded from analysis due to incomplete biomarker or clinical information, withdraw of informed consent or double entry (see patient flow diagram; figure 1). The final study cohort consisted of 2294 patients (57.2% males, median age 57 years) with suspected ACS. Numbers of recruited patients varied by study site but were limited per protocol to a maximum of 300 enrolments per site. The exact numbers of recruited patients are displayed in online supplementary figure 1S.
Supplemental material
The most prevalent leading symptom at presentation (online supplementary figure 2S, table 1) was chest pain in 70.6% (n=1619), followed by diffuse or initially mixed symptoms in 12.9% (n=297), dyspnoea in 5.2% (n=119), abdominal pain in 2.9% (n=66), focal neurology in 0.7% (n=16), headache in 0.4% (n=9) or none of the listed symptoms in 7.3% (n=168). As expected from the inclusion criteria, the study cohort represented a low-to-intermediate risk group with a median GRACE score of 89 (IQR: 67 to 114) and a Killip class of 1 in 96% of cases (n=2084). Time from onset of symptoms to presentation was below 12 hours in 50.8%. An interval of 0 to 3 hours, 3 to 6 hours and 6 to 12 hours was registered in 26.3% (n=558), 13.3% (n=283) and 11.2% (n=238) of patients, respectively. ECG at presentation was non-diagnostic in 87.3% of patients. Regarding initial cTn and Copeptin results, a total of 2017 patients (87.9%) were below the diagnostic cut-off of the local cTn, and 1615 patients (70.4%) below the cut-off for Copeptin. A total of 1477 patients (64.4%) were below the decision cut-off for both biomarkers fulfilling the criteria for early primary discharge from the ED (theoretically maximal efficiency).
Supplemental material
Clinical pathways
Nine hundred and seventy-four patients (42.5%) were categorised into the primary discharge after fast rule-out pathway, and 1320 patients into the conventional workup pathway. Of these, 654 patients did not follow a predefined pathway but were either admitted although qualified for primary discharge (n=503, 21.9%), or were discharged although not ruled out (n=151, 6.6%), see figure 2.

{kind=link}
![[bmjopen-2018-028311supp003.jpg]](https://bmjopen.bmj.com/content/bmjopen/9/7/e028311/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
![[bmjopen-2018-028311supp004.jpg]](https://bmjopen.bmj.com/content/bmjopen/9/7/e028311/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
Algorithm for an early rule-out strategy and guidance of primary early discharge versus general hospital admission (conventional workup). ACS, acute coronary syndrome.
In the entire cohort, the overall rate of an ACS diagnosis was 12.7% (n=288), non-cardiac chest pain 28.8%, rhythm disorders 8.7%, pulmonary disorders 6.8%, stable CAD 6.8%, hypertensive crisis 6.3% and gastrointestinal disease 5.5%. Other cardiac diagnoses were present in 4%, and other unspecified diagnoses in 16.3% of cases (online supplementary figure 3S).
Supplemental material
![[bmjopen-2018-028311supp005.jpg]](https://bmjopen.bmj.com/content/bmjopen/9/7/e028311/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
In the conventional care pathway, an ACS was diagnosed in 21.1% (n=279) with the majority classified as a NSTE-ACS (n=172, 61.6%). STEMI was an exceptional diagnosis in 15 patients (5.2%) since patients with STEMI were routed directly to the catheterisation laboratory in most institutions and were not intended for inclusion. Only if STEMI was diagnosed later and not at admission such patients were enrolled. Other diagnoses included non-cardiac chest pain in 18.8% (n=247), rhythm disorders in 5.9% (n=133), stable CAD in 8.9% (n=117), pulmonary disease in 6.8% (n=90), hypertensive crisis in 5.9% (n=77), gastrointestinal disease in 4.7% (n=62) and other diagnoses in 14.1% (n=185).
In the primary discharge after fast rule-out pathway, only nine patients (0.9%) were diagnosed as having an ACS, mostly unstable angina (n=4) or unclassified ACS (n=4), with only one case (0.1%) diagnosed as non-ST-segment elevation myocardial infarction (NSTEMI) (NPV for MI of 99.9%). Rate of admission was only 0.1% due to a case where admission was forced by the referring primary care physician although discharge was planned.
There were two different ways how local investigators over-ruled the intended pathway. The larger group consisted of 503 patients (21.9%) who were allocated to the conventional care pathway at the discretion of the local investigator although they were categorised into the primary discharge after fast rule-out pathway. The second group consisted of 151 patients (6.6%) who were primarily discharged although they should have received conventional care. Reasons for the over-rule consisted mainly of decisions of the physician to admit to hospital based on clinical judgement. Minor reasons were opposition of patients against serial blood sampling (n=2) and other unspecified reasons (n=6).
There were differences between the primary discharge after fast rule-out pathway and the over-rulers into the conventional care pathway (table 2). Patients were older, more frequently males, had more often a history of CAD or previous MI, more risk factors including a higher prevalence of arterial hypertension, hypercholesterolaemia and diabetes mellitus. In addition, patients had more often a diagnostic ECG, and higher GRACE scores. In addition, these patients received more often an ACS diagnosis, that is, a diagnosis of unstable angina, and spent longer times in the ED. However, and importantly, rates of all-cause mortality at 30 days were not significantly different (0.2% vs 0.1%, p=1) compared with the primary discharge after fast rule-out pathway.
Comparison of patient’s characteristics of primary discharge versus over-rule to conventional care despite eligibility for discharge by biomarker results
Outcomes
The primary endpoint, all-cause death within 30 days among the primary discharge after fast rule-out pathway, occurred in only one case of 974 patients (0.1%, 95% CI: 0% to 0.6%). This death was not related to the biomarker algorithm: the patient was 70 years old, had a history of CAD and previous MI and presented with musculoskeletal symptoms, was primarily discharged and died 1 month later from metastatic lung cancer (table 3).
All-cause death at 30 days and secondary outcomes
By contrast, all-cause mortality rate in the conventional care pathway was 1.1% (14 of 1320 patients, 95% CI: 0.6% to 1.8%) and thus significantly higher (p=0.011) than in the primary discharge after fast rule-out pathway (table 3). Diagnoses in the deceased patients of the conventional care pathway included ACS (n=5), non-cardiac chest pain (n=2), pulmonary disease (n=2), neurological disease (n=1), rhythm disorders (n=1), stable CAD (n=1), heart failure (n=1), gastrointestinal disease (n=1) and non-specified others (n=1). Patients who died were a median of 15 years older, had more often dyspnoea as the leading presenting symptom, presented more frequently more than 12 hours after symptom onset and were characterised by higher GRACE score (167 vs 90 points, p<0.001) and Killip class. In addition, non-survivors had received more extensive diagnostic workup, presented more often with a local cTn and Copeptin above cut-off and median Copeptin values were significantly higher than among survivors (50.8 vs 7.0 pmol/L, p<0.001) underscoring the prognostic information that is provided by cTn and Copeptin independent of the underlying disease.
Regarding secondary endpoints, hospitalisation rates were 0.1% in the primary discharge after fast rule-out pathway compared with 59% in the conventional care pathways (p<0.001). As expected, median lengths of stay in the ED (treatment time) were significantly shorter in the primary discharge after fast rule-out pathway versus the conventional care pathway (228 min vs 288 min, p<0.001) and rates of patients discharged within 0 to <1 hour (1.5% vs 3.6%), 1 to <2 hours (13.2% vs 13.3%), 2 to <3 hours (21.7% vs 16%), 3 to <6 hours (49.3% vs 37.3%) were significantly different in primary discharge after fast rule-out pathway versus conventional care pathway (p for trend <0.001). Conversely, rates of patients with longer ED treatment times >6 hours were significantly lower in the primary discharge after fast rule-out pathway than in the conventional care pathway out group (14.2% vs 29.8%, p<0.001).
Discussion
Information on the safety of direct discharge from an ED after rule-out of MI in patients with suspected ACS is almost exclusively restricted to findings that were generated in observational trials where attending physicians were commonly blinded to the investigational hs-cTn results, or to retrospectively determined optimal decision cut-offs. Treatment decisions based on at that time applicable standards of care and were left at the discretion of the treating physician.16–19 31
Following the randomised BIC-8 study, which proofed safe discharge after instant rule-out of AMI by the use of troponin and Copeptin from a single blood draw22 and also showed cost-effectiveness in a health economic substudy,32 we could confirm in a large European registry that this is also true in clinical routine.
The superior analytical sensitivity of hs-cTn assays has already enabled an accurate rule-out of MI with sensitivities and NPVs of >90%,10 facilitating fast rule-out based on either very low concentrations of hs-cTn assays obtained from a single measurement at presentation,14–19 33 or from serial blood draws after 1 to 3 hours17–19 31 34–39 using hs-cTn at the 99th percentile10–13 or slightly below18 19 the 99th percentile of a healthy reference population. Integration of clinical judgement or a validated clinical score such as the GRACE, TIMI, HEART, modified Goldman Score, MACS clinical decision rule, EDACS and Vancouver Chest Pain Algorithm and North American Chest Pain Rule further improve NPV yielding NPV between 98.1% to 100% and 98.4% to 100% when cTn and hs-cTn assays were used, respectively.40 Although, 2015 ESC guidelines10 discourage routine invasive strategy in low risk patients and rather recommend discharge following risk stratification, and a pre-discharge or post-discharge stress imaging test to decide on a selective invasive strategy, evidence from randomised trials to endorse these recommendations is sparse.20–22 The Manchester Acute Coronary Syndrome (MACS)-Pilot study20 enrolled 138 patients with suspected cardiac chest pain who were randomised to receive care guided by the MACS decision rule or standard care. The primary efficacy outcome was a decision to discharge within 4 hours of arrival, without missed MI and without death, AMI or coronary revascularisation occurring during 30 days of follow-up. This small pilot study found a significantly higher rate of uneventful primary discharge within 4 hours (26% vs 8%, p=0.004) among those guided by the MACS rule. The HEART Pathway Trial enrolled 282 patients with suspected ACS stratified into risk categories using the HEART Score.21 The study was not powered to compare event rates in randomised groups but found a decreased objective cardiac testing at 30 days by 12.1%, a reduced length of stay by 12 hours and an increase of early discharges by 21.3%. The BIC-8 trial22 that enrolled a total of 902 low-to-intermediate high risk patients using the GRACE score and subsequently randomised patients with normal presenting cTn and Copeptin values into an early discharge and a standard protocol group. The study demonstrated a reduction of observation time in the ED by more than 40% from a median of 7 hours to 3 hours, achieved a 5.6-fold increase in ED discharge rate from 67.7% versus 12%, and a similar 5.2% rate of 30 day major adverse cardiovascular events that were liberally defined as all-cause death, survived sudden cardiac arrest, re-hospitalisation for ACS, unplanned PCI or CABG or documented life-threatening arrhythmias in the standard and Copeptin group.22
The present large multicenter registry was performed in patients with suspected ACS and low-to-intermediate risk to test the usefulness of a dual biomarker strategy, consisting of a normal Copeptin and cTn, to rule-out MI from a single blood draw at admission and to discharge low risk patients primarily from the ED. In order to represent clinical practice of different type of institutions, variable local practice and across the spectrum of cTn assays and grades of assays sensitivities,41 42 this observational study was conducted in 18 different institutions in Europe and Asia. Institutions included EDs in community hospitals, and CPUs in PCI centres and few university hospitals. Patients qualified for enrolment in the presence of a broader spectrum of symptoms suggestive of ACS not limited to chest pain or angina, and a broad spectrum of cTn assays and different grades of analytical sensitivities including conventional, contemporary and hs-cTn assays was permitted. To reduce dominance of few high recruiting centres, enrolment rates were restricted to 300 study patients per site.
There were several key findings of this survey that support the usefulness and safety of this concept in clinical routine and outside of controlled clinical trials. First, earlier discharge from the ED in patients ruled-out at presentation using a single blood draw is feasible without any obvious safety concern. All-cause mortality rate within 30 days was 0.1% and attributed to a case with metastatic lung cancer. Second, length of stay in the ED is significantly shorter by 60 min allowing an earlier discharge, a finding particularly useful in congested EDs or CPUs. Thus, the present registry data confirm the findings from the randomised BIC-8 trial22 on reduced length of stay, increased discharge rates and support the safety of a primary planned discharge from an ED after clinical risk assessment. Third, the dual marker concept is efficient as it can be applied to at least 42.5% (potentially effective in 66.4%) of patients presenting with chest pain or chest pain equivalent symptoms to an ED. Thus, efficacy of this dual marker strategy is almost comparable with the efficacy of the ESC recommended 0/1 hour diagnostic algorithm that requires serial blood draws and a validated hs-cTn assay (currently Abbott Architect hs-cTnI and Roche hs-cTnT). While other fast rule-out algorithms based on very low hs-cTnI or hs-cTnT at the limit of blank or limit of detection may demonstrate similar diagnostic performance and safety, the numbers of patients who qualify are substantially lower14 15 33 and these strategies have never been tested prospectively with patients being really discharged after testing.
We found a relevant number of over-rule by local ED physician leading to an admission of patients who qualified for discharge by their biomarker results (34%). Given that these patients had an uneventful clinical course (see table 2), void of primary or secondary events during follow-up, suggests an underestimated efficacy and more potential of safe discharge. Fourth, regarding the diagnostic performance for rule-out that was not in the scope of this survey, the dual marker algorithm was associated with a high negative predictive value of 99.9% for NSTEMI (one missed NSTEMI) confirming the existing evidence on the diagnostic performance of the Copeptin/troponin dual marker strategy.22 26–28 Fifth, regarding secondary objectives, the dual marker strategy was associated with shorter stays in ED. Sixth, consistently with previous studies,26–28 43 44 elevated Copeptin levels were associated with all-cause mortality within 30 days providing confirmatory evidence that Copeptin confers prognostic information that is complementary to cTn or hs-cTn, in various acute cardiovascular settings including ACS,26–28 43 44 heart failure45 46 and acute pulmonary embolism47 but also non-cardiac disease. In addition, an elevated Copeptin should prompt a search for a variety of potentially life-threatening non-cardiac conditions including perforated stomach ulcer, pancreatitis, cholecystitis, bleedings, infections or neurological disorders.48
Limitations
First, we observed very low rates of all-cause mortality at 30 days, that is, 0.1% (95% CI: 0% to 0.6%) in the primary discharge after fast rule-out pathway as compared with 1.1% (95% CI: 0.6% to 1.8%) in the conventional care pathway. Low event rates may be explained by restriction of the DMS algorithm to patients at low or intermediate risk based on the GRACE score. Therefore, our findings cannot be extrapolated to settings where risk stratification after rule-out is based on other clinical scores or on clinical judgement. Moreover, a selection bias towards recruitment of a non-representable low risk ACS cohort cannot be fully excluded as inclusion criteria were not limited to typical chest pain, longer pain episodes or abnormal ECG findings. However, the study population was planned to represent a real life picture of patients who present in clinical routine with various symptoms and a wide range of risk. Copeptin concentration return to normal within few hours reducing the diagnostic performance of the DMS algorithm to early presenters. As a tribute to the consecutive enrolment of patients, we were not able to enrich the study population by patients presenting within 6 hours from onset of symptoms (49.2% of the entire study cohort reported onset of symptoms more than 12 hours before presentation). Therefore, scrutiny is advised regarding the interpretation of the DMS result in patients presenting very late or who cannot state a precise onset of symptoms. We believe that our study cohort is also similar to other observational studies enrolling patients with suspected ACS. The overall prevalence of ACS in this registry was 12.7% and is thus very consistent with a median of 13% to 14% prevalence of ACS reported in a pooled analysis of 51 observational trials on patients with suspected ACS.2 In addition, the median GRACE score was 89 points (IQR: 67 to 114) which is very similar with the mean GRACE score of 80 (SD 28 points) in the randomised intervention trial.22
Second, rates of enrolment per site were heterogeneous with a mix of high and low recruiting centres. However, the very low mortality rate does not allow any exploratory analyses on the safety of discharge by centre volumes, experience of physicians, local cTn assay or assay generation.
Third, currently a strategy for instant rule-out based on Copeptin and cTn is being recommended by 2015 ESC guidelines on NSTE-ACS10 and an updated consensus document of the German Society of Cardiology on the use of Copeptin in CPUs49 and chest pain centres.50 However, there is a gap between the high recommendation level endorsed by numerous clinical trials,23–26 43 44 editorials and state-of-the-art reviews,38 40 meta-analysesp27 28 and national practice guidelines10 49 50 on the one hand and the obvious underuse in clinical practice for suspected ACS. In the elective setting, Copeptin is currently used for the diagnosis of diabetes insipidus, a non-emergent diagnosis. In emergencies requiring immediate measurement, the most probable reason for underuse is that Copeptin has to be measured on a stand-alone device that is more labour-intensive than an automated central laboratory system, which leads to the suspicion that nowadays economic features in the laboratory are hurdles for state of the art use of biomarkers. Development of a POCT system for Copeptin and implementation of Copeptin to a central laboratory platform would overcome this obstacle. In this registry, however, Copeptin was measured on a Kryptor platform with a measuring time of 14 min and immediate reporting of the result to the ED physician. Accordingly, most of the time delays between diagnosis and the disproportionally longer stay in ED are regarded to be related to other time consuming processes including diagnostic workup for differential diagnoses and drafting of the discharge report, particularly in the presence of crowding in the ED.
Conclusions
Copeptin on top of cardiac troponin is currently the only strategy that – based on a randomised control trial and a large multicentre registry - supports the safe direct discharge of patients with chest pain or chest pain equivalent symptoms suggestive of ACS under routine conditions. There are only few randomised trials that provide evidence for a safe discharge after rule-out in low risk patients. The present registry confirms findings from the randomised BIC-8 trial in an independent real world registry. The efficacy of the DMS in terms of patients potentially qualifying is at least 42.5% or potentially considerably higher.
We believe that the present findings have potential impact on healthcare resources by shortening observation times, hospitalisation rates, reducing diagnostic resources and avoid unnecessary coronary angiographies should barriers to adoption be overcome.
Acknowledgments
We want also to acknowledge the participant centres and their local contributors for the support in data collection and for providing results in a timely manner (in alphabetical order by country): Universitätsklinikum Tulln, Tulln (Austria): Keywan Bayegan, Herbert Frank. Wilhelminenspital, Vienna (Austria): Alja Gomiscek, Kurt Huber, Mona Kassem, Kris Vargas. Centre Hospitalier de Calais, Calais (France): Anthony Nghi. Hôpital du Bocage - CHU, Dijon (France): Didier Honnart. Centre Hospitalier Universitaire de Montpellier, Montpellier (France): Anne-Marie Dupuy, Sophie Lefebvre, Mustapha Sebbane. Kerckhoff-Klinik, Bad-Nauheim (Germany): Christian Hamm, Christoph Liebetrau. Berlin Hedwigshöhe, Berlin (Germany): Malte Schröder. Charité – Universitätsmedizin Berlin, Berlin (Germany): Kim Kastner, Martin Möckel, Anna Slagman. BG Klinikum Marzahn, Berlin (Germany): Berthold Hoppe, Hinrich Schroer, Susann Schweitzer, Mirko Seidel. St. Elisabeth Krankenhaus, Mayen (Germany): Katja Bininda, Michael Maasberg, Ralph Rüdelstein. Budapest Semmelweis University, Budapest (Hungary): Peter Kanizsai. Vilnius University Hospital Santariškių Klinikos, Vilnius (Lithuania): Renata Ruseckaite, Pranas Serpytis. Kantonsspital Aarau, Aarau (Switzerland): Ulrich Bürgi. Spital Zollikerberg, Zollikerberg - Kanton Zürich (Switzerland): Thomas Gaisl. Spital Zollikerberg, Zollikerberg - Kanton Zürich (Switzerland): Thomas Gaisl. Hacettepe University, Ankara (Turkey): Zeliha Günnur Dikmen. Bucks Healthcare Wycombe Hospital, High Wycombe (UK): Nicola Bowers, Piers Clifford, Josephine Chaplin, Mari Kononen, Anu Maharajan
References
Footnotes
Contributors EG and MM were involved in the conception and design of the study, the acquisition, analysis and interpretation of data, drafted the manuscript, approved the final version to be published, are accountable for all aspects of the work and and MM serves as guarantor for the manuscript. CS, JOV, CL, RR, AS, HS, MM-H, MS, DH, KH, CH and PC were involved in the interpretation of data, critically revised the manuscript for important intellectual content, approved the final version to be published and agreed to be accountable for all aspects of the work. KK was involved in the interpretation and management of data, critically revised the manuscript for important intellectual content, approved the final version to be published and agreed to be accountable for all aspects of the work. JCW was involved in the interpretation and statistical analysis of data, critically revised the manuscript for important intellectual content, approved the final version to be published and agreed to be accountable for all aspects of the work. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Funding This work is an investigator initiated analysis and was financially supported by Thermo Fisher Scientific BRAHMS GmbH (https://www.brahms.de). The funders had no role in study design, data collection and specification of statistical analysis, decision to publish or preparation of the manuscript. Note that the implementation of statistical analysis was conducted by BRAHMS.
Competing interests EG received honoraria for lectures from Roche Diagnostics, AstraZeneca, Bayer, Daiichi-Sankyo, Lilly Eli Deutschland. He serves as a consultant for Roche Diagnostics, BRAHMS Thermo Fisher Scientific, Boehringer Ingelheim and has received research funding from BRAHMS Thermo Fisher Scientific, Roche Diagnostics, Bayer Vital and Daiichi Sankyo; AS has received research funding from Roche Diagnostics, BRAHMS Thermofisher Scientific and the German Research Council (DFG); MM received honoraria for lectures from Roche Diagnostics, AstraZeneca, Bayer Vital, Daiichi-Sankyo, Boehringer Ingelheim and BRAHMS Thermo Fisher Scientific. He serves as a consultant for BRAHMS Thermo Fisher Scientific and Bayer, and has received research funding from BRAHMS Thermo Fisher Scientific, Roche Diagnostics and Radiometer. CS, JOV, JCW are employees of BRAHMS Thermo Fisher Scientific. MM-H report research grants from Roche Diagnostics, BRAHMS Thermo Fisher Scientific and the University of Heidelberg. Speaker honoraria from RocheDiagnostics KK reports fees from BRAHMS Thermo Fisher Scientific for monitoring activities related to the study. DH reports speakers fees from BRAHMS Thermo Fisher Scientific. KH received honoraria for lectures from AstraZeneca, Bayer, Boehringer Ingelheim, BRAHMS Thermo Fisher Scientific, Daiichi Sankyo, Pfizer, Sanofi and The Medicines Company and has received research funding form AstraZeneca and BRAHMS Thermo Fisher, respectively. ChH and CPC report speakers fees and honoraria for consultancy from BRAHMS Thermo Fisher Scientific.
Ethics approval The study received the primary ethics approval from the Charité (“Ethikausschuss 1am Campus Charité-Mitte; EA1/008/15).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data available.
Patient consent for publication Not required.