Article Text

Abstract
Introduction Respiratory failure requiring endotracheal intubation accounts for a significant proportion of intensive care unit (ICU) admissions. Little attention has been paid to the laryngeal consequences of endotracheal intubation. Acute laryngeal injury (ALgI) after intubation occurs at the mucosal interface of the endotracheal tube and posterior larynx and although not immediately manifest at extubation, can progress to mature fibrosis, restricted glottic mobility and clinically significant ventilatory impairment. A recent prospective observational study has shown that >50% of patients intubated >24 hours in an ICU develop ALgI. Strikingly, patients with AlgI manifest significantly worse subjective breathing at 12 weeks. Current ALgI treatments are largely surgical yet offer a marginal improvement in symptoms. In this study, we will examine the ability of a postextubation medical regime (azithromycin and inhaled budesonide) to improve breathing 12 weeks after ALgI.
Methods and analysis A prospective, single-centre, double-blinded, randomised, control trial will be conducted at Vanderbilt Medical Center. Participants will be recruited from adult patients in ICUs. Participants will undergo a bedside flexible nasolaryngoscopy for the identification of ALgI within 72 hours postextubation. In addition, participants will be asked to complete peak expiratory flow measurements immediately postintubation. Patients found to have ALgI will be randomised to the placebo control or medical therapy group (azithromycin 250 mg and budesonide 0.5 mg for 14 days). Repeat peak expiratory flow, examination of the larynx and patient-reported Clinical COPD (chronic obstructive pulmonary disease) Questionnaire, Voice Handicap Index and 12-Item Short Form Health Survey questionnaires will be conducted at 12 weeks postextubation. Consented patients will also have patient-specific, disease-specific and procedure-specific covariates abstracted from their medical record.
Ethics and dissemination The Institutional Review Board (IRB) Committee of the Vanderbilt University Medical Center has approved this protocol (IRB #171066). The findings of the trial will be disseminated through peer-reviewed journals, national and international conferences.
Trial registration number NCT03250975
- acute laryngeal Injury
- medical management
- laryngotracheal stenosis
- post-icu dyspnea
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Randomised trial to investigate the efficacy of pharmacologic management of acute laryngeal injury after endotracheal intubation.
This study offers a novel treatment paradigm to reduce the burden of post-intensive care unit dyspnoea.
Primary endpoints will include anatomic, physiologic and patient-reported outcomes.
Although generalisability of the results of this phase II study may be limited given its single-centre setting, the outcomes will inform a multicentre phase III trial.
Introduction
Background and rationale
Fifty-five thousand patients are treated in intensive care units (ICUs) every day in the USA. Respiratory failure requiring mechanical ventilation is the leading indication in the ICU admissions.1 Although most patients survive, many develop persistent disabilities that limit the return to their preadmission function. Although a recent study has centred on the constellation of cognitive,2 psychological3 and physical impairments that arise after treatment in an ICU, little attention has been paid to the laryngeal consequences of endotracheal intubation.
Laryngeal injury after intubation occurs at the mucosal interface of the endotracheal tube and posterior glottis.4–6 Animal models of laryngeal injury show rapid infiltration of inflammatory cells, neovascularised granulation tissue7 and subsequent weakening of airway cartilage.8 Although not immediately manifest at extubation, mucosal injury can progress to mature fibrosis, restricted glottic mobility and ventilation impairment that significantly impact the quality of life.9 A prospective observational study demonstrated that of 61 patients, 41% had vocal fold immobility. Patients who were intubated for >48 hours underwent flexible nasolaryngoscopy after extubation. The laryngeal injury was defined as granulation tissue, ulcerations and vocal fold immobility.10 Oversized endotracheal tubes are significantly associated with an increased risk of acute laryngeal injury (AlgI) due to greater tracheal wall pressure leading to more severe injury.8 However, a majority of patients with ALgI have a properly sized endotracheal tube (ETT) but possesses intrinsic risks factors that predisposed them to disease development.
Current ALgI treatments occur when patients present far after ICU discharge. Treatment is primarily surgical and offers marginal improvements in ventilation yet is associated with a significantly worse voice. Preventative efforts to preserve laryngeal function through early intervention could reduce the incidence of this devastating complication of medical care11 and avoid invasive open surgical reconstruction.12 Preliminary data suggest early identification of ALgI after intubation may prevent the development of chronic complications. Patients with early interventions for postintubation tracheal injury required fewer total interventions, had longer intervention free intervals and did not require external laryngotracheal reconstruction in comparison with patients treated for chronic fibrotic scars.13
The development of airway fibrosis and anatomic ventilatory obstruction is associated with fibroblast proliferation and deposition of the extracellular matrix, in particular, collagen types I and III,14–16 transforming growth factor- β1 (TGF- β1) and interleukin-17 (IL-17).17 Tracheal fibroblasts showed elevated TGF-β, COL1A1 and COL3A1 in patients with laryngotracheal stenosis.18 IL-17 is linked to the extracellular matrix deposition leading to fibrotic disease and has a role in idiopathic subglottic stenosis.17 19 Based on logical progression, it follows that anti-inflammatory agents may play a role in mediating mucosal airway inflammation. Macrolides, in particular, have been recognised to have anti-inflammatory effects beyond their antimicrobial properties. Azithromycin has been shown to significantly reduce nuclear factor-kappa B activation and the synthesis of pro-inflammatory cytokines in tracheal aspirate cells.20 A recent study investigating drug combinations for the prevention and treatment of tracheal stenosis in a rabbit model found that erythromycin, combined with budesonide, may reduce inflammation and modify the disease process of tracheal stenosis after tracheal injury.21 In addition, another study demonstrated that erythromycin combined with penicillin and budesonide were most effective in inhibiting the proliferation of granulation tissue after tracheal injury.22 23 Azithromycin has been shown in the chronic obstructive pulmonary disease (COPD) population to reduce exacerbations and improve quality of life.24 Furthermore, a case study demonstrated the positive effects of the combination of azithromycin and prednisone on subglottic stenosis.23
Patients with endotracheal intubation are at risk for laryngotracheal stenosis, and yet current therapies are unsatisfactory. Preliminary investigations of drug combinations have shown promise at arresting the natural history of the disease and improving patient outcomes. In this study, we will examine the ability of a postextubation medical regimen (azithromycin and budesonide) to improve breathing 12 weeks after ALgI.
Objectives
Initiate a randomised control trial to investigate the ability of azithromycin and budesonide to improve objective and subjective breathing measures in patients with ALgI following endotracheal intubation.
Trial design
A two-arm randomised control trial. Participants and their providers will not be aware of their status as control or being in the drug group (figure 1). This randomised approach will enable us to compare the clinical effectiveness of the medical regime in improving 12-week primary endpoints of 1. Clinical dyspnoea (via participant reported Clinical COPD Questionnaire, CCQ scores). Secondary endpoints include ventilatory restriction (via peak expiratory flow rate) and anatomic injury (restricted vocal fold mobility over intake baseline).

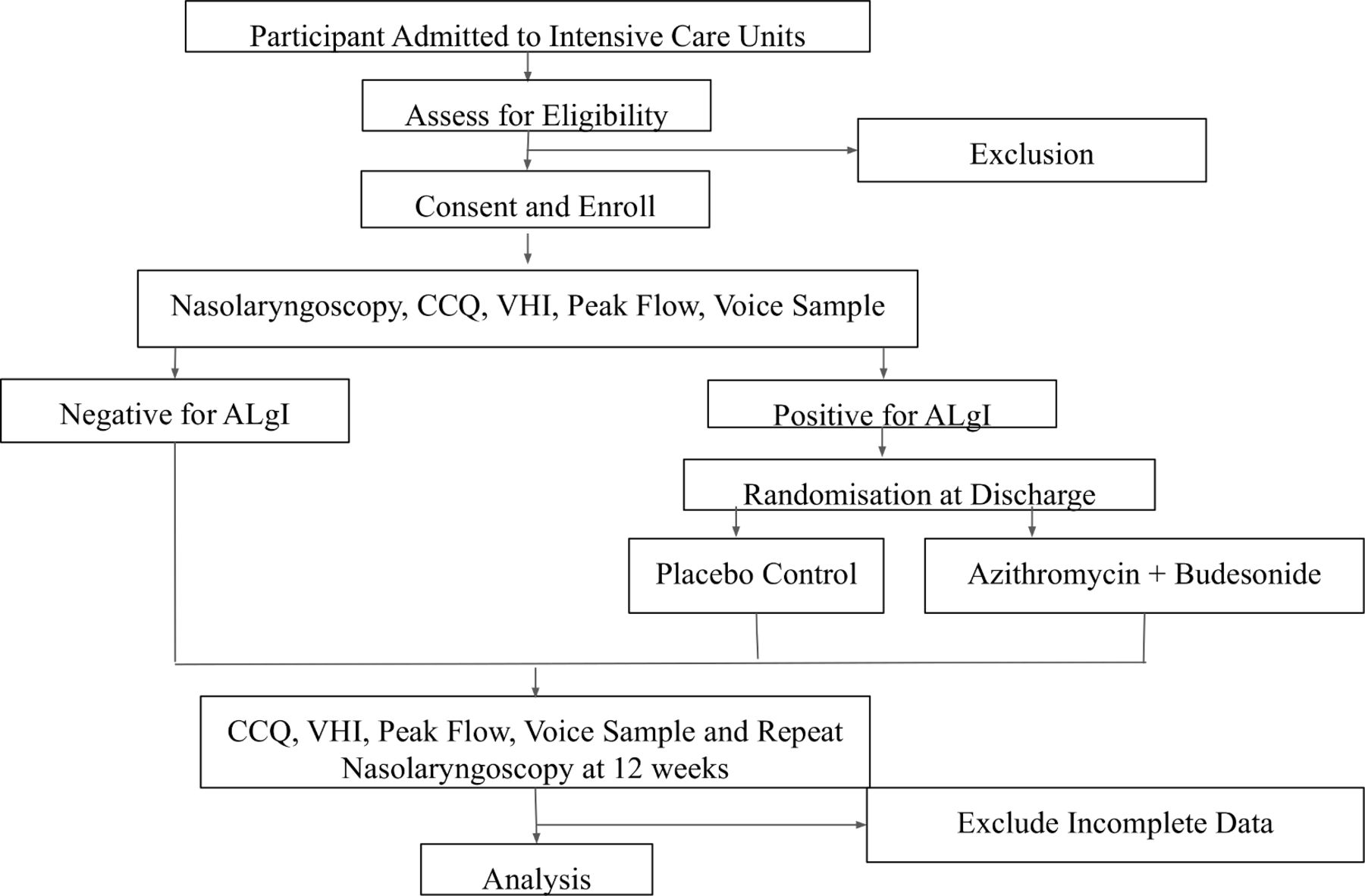{kind=link}
Schematic overview of anticipated trial work flow. CCQ, Clinical COPD (chronic obstructive pulmonary disease) Questionnaire; VHI, Voice Handicap Index.
Methods: participants, intervention and outcomes
Study setting
A prospective, single-centre, double-blinded, randomised controlled trial will be conducted at Vanderbilt University Medical Centre (VUMC). Vanderbilt Medical Center is a tertiary care centre several high-volume critical care units (including a Medical ICU, Surgical ICU, Trauma ICU and Burn ICU), as well as a centre for complex laryngeal and tracheal disease (Vanderbilt Center for Complex Airway Reconstruction).
Adult patients in a VUMC ICU who have undergone >24 hours but <7 days of endotracheal intubation will be eligible. Patients with previous incidences of prolonged intubations defined as >24 hours will be excluded due to the inability to adequately discern patient breathing and voice outcomes from past versus present injury. Patients with an intubation period of >7 days will be excluded to keep the recruited patient population homogeneous in terms of potential laryngeal injury. Potential candidates will be approached for participation in the study within 72 hours of extubation by the study investigator.
Eligibility criteria
Inclusion criteria: Adult participants with >24 hours and <7 days of intubation in an ICU.
Exclusion criteria:
Age under 18 years on admission.
Patients with anticipated discharge 5 days after extubation.
Patients who are dependent for activities of daily living in the 30 days prior to admission.
Patients unable to consent.
Patients with neck trauma.
Patients with head and neck malignancies.
Patients with pre-existing laryngeal or tracheal stenosis.
Patients with other pre-existing respiratory conditions such as neuromuscular dystrophies, cystic fibrosis, bronchiectasis.
Patients who had been previously intubated for an extended period of time.
Patients who are pregnant or currently breastfeeding.
Patients with allergies to study medications.
Patients with a resting heart rate >100 beats per minute.
Patients with a prolonged corrected QT (QTc) interval (>450 msec) or the use of medications that prolong the QTc interval or are associated with Torsades de pointes (with the exception of amiodarone).24
Patients with severe hearing impairment documented by audiometric testing.
Interventions
Participants will be recruited from adult patients in the surgical and medical ICUs of Vanderbilt Medical Center. Following an assessment of study eligibility, informed consent and study enrollment, patients will undergo a routine medical examination of their larynx with flexible nasolaryngoscopy after anaesthetising and decongesting the nose with 1% lidocaine mixed in oxymetazoline. During the examination, patients will be asked to sniff (which triggers the opening of the vocal cords), and say ‘Eee’ which (triggers closing). These manoeuvres allow assessment of vocal fold movement. The evaluation will be recorded for subsequent blinded review. Recordings will be coded with a research identifier and stored digitally and securely in a RedCap database. Participants will also produce a digital voice samples and produce a peak flow measurement (average of three attempts via a peak flow metre), as well as provide a voice sample through a REDCap recording.
The presence of acute laryngeal injury will be documented in a binary fashion (presence or absence). Additionally, the physical examination findings of the larynx will be explicitly delineated on the standardised REDCap study evaluation form.
The participant with acute laryngeal injury will be randomised at discharge to either a non-drug placebo control group or a medical therapy group consisting of azithromycin 250 mg and budesonide 0.5 mg for 14 days. Participants will receive a communication via telephone or text message weekly or bi-weekly to encourage compliance with medication. Monthly, patients will be asked to complete a CCQ over the phone or through an electronic survey captured in a secure REDCap database.
For all participants, a repeat office-based examination of the larynx will be scheduled at 12 weeks. At that time, participants will also be asked to complete the CCQ,25 Voice Handicap Index (VHI),26 12-Item Short Form Health Survey, (SF-12),27 questionnaires and produce an average peak flow measurement after three attempts via a peak flow metre and provide a voice sample through a REDCap recording. The CCQ and the VHI are validated and sensitive instruments for assessing symptom severity in patients with laryngotracheal stenosis.13 28
Consented patients will also have information abstracted from their medical record. Pertinent information collected will include patient-specific covariates; that is, medical comorbidities, laboratory values and demographics as well as procedure-specific covariates; that is, endotracheal tube size, length of intubation, recorded endotracheal tube cuff pressures, hospital location of intubation, intubating specialty and the number of recorded intubation attempts. The status of laryngeal injury will be graded by two blinded otolaryngologists independently. If the two reviewers differ in their assessment, a third blinded otolaryngologist will be invited to make the classification.
The initial and follow-up examinations will be non-billable and provided no cost to the patients. Data will be stored in a secure REDCap database housed within a server located behind the VUMC firewall. Data analysis will be performed by Key Study Personnel (KSP) at VUMC.
Outcomes
Primary outcome measures:
12-week patient-reported dyspnoea via CCQ.25
Secondary endpoints
12-week assessment of laryngeal function using flexible nasolaryngoscopy.
12-week patient-reported voice via VHI-10.26
Exploratory Outcomes
12-week patient peak expiratory flow rate (via handheld peak flow metre).
Quality of voice, as measured by the Grade, Roughness, Breathiness, Asthenia, Strain (GRBAS) scale.29
Overall quality of life, as measured by SF-12.27
Readmission, including those for non-laryngeal causes.
30-day all-cause mortality.
Adverse events, subdivided into suspected unexpected serious adverse reactions, serious adverse reactions, and adverse reaction.
Persistent tracheostomy at 12 weeks.
Participant timeline
Within 12–72 hours of extubation, participants will be approached to enrol in the study. Once the informed consent is signed, the patient will complete the questionnaires, provide a peak flow and undergo the flexible nasolaryngoscopy. Prior to discharge, the patients will be randomised to either the placebo control or medical therapy group. Participants will be educated on the medications by a pharmacist. They will then take the azithromycin 250 mg and budesonide 0.5 mg 14 days. A KSP will get in contact with the participants weekly to ensure completion of the drug regimen and to remind the patient about the follow-up components. At 12 weeks, the patients will be asked to return to follow-up clinic, complete the questionnaires, provide a peak flow and provide a voice sample. They will also undergo a second flexible nasolaryngoscopy at this time. Patients identified to have chronic laryngeal injury will be encouraged to follow-up at the Voice Clinic at Vanderbilt Medical Center.
Sample size
We are planning a study of a continuous response variable from independent control and experimental subjects. To calculate sample size, we have used CCQ data in a previous study, in which the response within each subject group was normally distributed with SD of 11.25. If the true difference in the experimental and control means is 9.15, we will need to study 24 experimental subjects and 24 control subjects to be able to reject the null hypothesis that the population means of the experimental and control groups are equal with probability (power) 0.8 (total n=48). The type I error probability associated with this test of this null hypothesis is 0.05. Anticipating 25% dropout rate, we will recruit 64 participants.
Recruitment
Patients who are eligible for the study will receive an oral explanation about the study from KSP. They will then be asked to sign a consent form on paper or electronically. A copy of the consent form will be put in their medical record. In the process of consenting patient, KSPs will stress that participants can withdraw from the study whenever they wish without giving a reason and without affecting their care in any way. If reasons were given, they will be documented.
Methods: assignment of interventions
Allocation
We will allocate all enrolled patients with ALgI at random between the placebo control group and the drug regime (budesonide and azithromycin group) at current clinical dosing. An independent pharmacist will randomly assign the patients (1:1), via a computer-generated randomisation sequence with permuted blocks of six (three per treatment group), to receive azithromycin 250 mg and budesonide 0.5 mg for 14 days or placebo pill and inhaled placebo for 14 days. Randomisation will be stratified by ALgI status (mild vs moderate/severe), then further stratified based on COPD status, creating four strata. The randomisation list will be retained by the clinical trials pharmacists at the Investigation Drug Services (IDS) at VUMC. The active medications and placebos will be prepared and packaged identically by a compounding chemist and dispensed by the IDS pharmacists according to the randomisation.
Blinding
All key personnel associated with the study will be blinded to the randomisation, except for the pharmacists until primary analyses are complete. Patients will be blinded to their treatment group assignment. In the case of adverse reactions to medications, participants will be unblinding is permissible.
Methods: data collection, management and analysis
Data collection methods
Laryngeal injury status will be digitally recorded using a bedside nasolaryngoscope utilising established criteria. Enrolled participants will also complete questionnaires (CCQ,25 VHI,26 SF-12,27 generate a peak flow measurement and provide a digital voice sample at baseline (within 72 hours of extubation) and at 12 weeks postextubation.
We will collect baseline data for all participants at recruitment, including:
Patient-specific variables
Age, sex, ethnic group, height, weight and BMI.
Contact information.
Disease-specific variables
Admission details, including diagnosis prompting intubation.
Active admission medical diagnosis.
Pre-existing medical comorbidities, in particular cardiorespiratory, liver, renal, diabetes and hypertension.
Admission laboratory values (RDW, Cr and CRP).
Markers of poor tissue perfusion (documented vasopressor requirement and sustained periods of hypotension).
Smoking status (active, historic and pack/years).
Procedure-specific variables
ETT size.
Length of intubation.
Recorded ETT cuff pressure.
Number of recorded intubation attempts.
Pre-intubation assessment (ie, Mallampati, grade of view and so on).
Data management
All data will be entered into an encrypted secure, password protected REDCap database. Data will be entered by an Institutional Review Board (IRB) certified physician or member of KSP. Only the principal investigator and co-investigator or key-study personnel will have access to the database.
We will have a consistent approach to missing data. Participants without any follow-up data will be excluded. For each variable, we shall summarise the frequency of missing data, which affects effective sample size and hence statistical power. If there is a reason to suspect that data are not missing completely at random, the trial statistician and chief investigator will discuss the findings. If not, we shall use appropriate imputation methods to ameliorate the problem of missing data
Statistical methods
We will first perform an intention-to-treat analysis, which will include all randomised participants based on their allocation, and use multiple imputation methods to account for missing data. After the intention-to-treat analysis is complete, we will also perform a per-protocol analysis, which will include participants who were completely adherent to their allocation group.
Baseline characteristics will be summarised by treatment groups. The patient characteristics, by treatment group, will be presented as mean (SD), median (25th–75th centile), minimum, maximum and count (%) depending on type and distribution. The significance level is set at 0.05 and all hypothesis testing will be two-sided.
The primary outcome, CCQ scores, will be presented using means and SD and the difference between the two groups will be presented as the difference in means with 95% CI. We will formally assess the distribution of the data for evidence of departure from normality. The Student t test or the Mann-Whitney U test will be used to compare the two study groups based on the distribution of the data. In the subsequent analysis, we will adjust for the following variables which may affect the degree of ALgI: ETT size, length of intubation, BMI, Charlson Comorbidity Index and diabetes mellitus type 2. A 5% level of significance will be used.
Continuous secondary and experimental outcomes will be analysed in the same fashion. Categorical secondary and experimental outcomes will be compared using the χ2 test. A 30-day all-cause mortality will be assessed using the Kaplan-Meier survivorship curve.
Methods: monitoring
Data monitoring
Responsibility for ensuring the reporting of adverse events (AEs) in accordance with the clinical trial regulations is delegated to the research team. All AEs are recorded and assessed by the PI or other designated person using an AE Screening Form to judge the seriousness, causality and expectedness of the event. Serious AEs are reported to the IRB Board at Vanderbilt.
Harms
The risk of release of protected health information exposure is present but will be limited with strict precautions. The nasolaryngoscopy poses minimal risk to the patient, as an endoscopic examination of the larynx is a minimally invasive procedure performed at the bedside or in a clinic room with the patient awake and alert. This examination is a routine portion of the medical examination performed on all patients undergoing evaluation of their airway after extubation, or when presenting to the outpatient clinic for voice evaluation. The limited risks from the examination include discomfort or mucosal irritation and trauma leading to epistaxis.
Normal clinical dosing for the medications will be used. Common side effects of budesonide include sore throat, congestion, epistaxis, oral thrush, cough, nausea, vomiting, diarrhoea and headaches. Rare but serious side effects of budesonide include but are not limited to increased risks of infection (ie, ear infections and pneumonia) fatigue, visual changes, mood changes, myalgia and slow wound healing. Common side effects of azithromycin include diarrhoea, nausea, vomiting abdominal pain, constipation, fatigue, headaches and dizziness. Rare but serious side effects of azithromycin include but are not limited to cardiac arrhythmias, liver dysfunction, increased cardiovascular and all-cause mortality. Due to these potential cardiovascular events, patients with previously recorded prolonged QTc intervals.
Auditing
Biannual internal audits of data integrity and completeness will be performed. An interim analysis will be performed at 6 months.
Patient and public involvement
Patients and the public were not directly consulted in the development of the research question or outcome measures. Patients were not involved in the recruitment and data collection process. The burden of intervention will not be assessed by trial participants. At the completion of this trial, a manuscript will be prepared to present the trial results. Results of the final study will be summarised and disseminated to all participants through their preferred method of communication indicated at the time of enrollment.
Ethics and dissemination
Research ethics approval
This study is approved by the IRB at Vanderbilt. IRB 171066.
Protocol amendments
Any protocol amendments will be communicated with the IRB and then communicated to the participants.
Consent
KSPs will obtain informed consent from the participants. A copy of the informed consent will be given to the participant for their record.
Confidentiality
All participant information will be stored within a secured server and will be collected, shared and maintained to protect confidentiality.
Declaration of interests
None declared.
Access to data
With the approval of the Vanderbilt IRB and the Principal investigator (PI), study documentation will be shared with requesting researchers and peer-reviewed journals to promote transparency and reproducibility in research.
Ancillary and post-trial care
Participants identified to have chronic laryngeal injury will be encouraged to follow-up with the Vanderbilt Otolaryngology department for post-trial care.
Dissemination policy
Results from this study will be pushed within the academic journal and presented at national meetings.
Acknowledgments
The authors would like to express their sincere gratitude to the Vanderbilt Institute for Clinical and Translation Research.
References
Footnotes
Contributors ASL, KK, JS, CS, AG: conceived the study, contributed to the study design and analytical plans. ASL and AG: drafted the protocol. All authors read and approved the final protocol.
Funding This project is funded through the NIH National Center for Advancing Translational Sciences: UL1 TR00224.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.