Article Text

Abstract
Introduction The common cold is the most common infectious disease affecting humans. It is usually a self-limiting disease; however, the common cold can cause significant morbidity and has a substantial economic impact on society. Human rhinoviruses (HRVs), which cause up to two-thirds of colds, have temperature-dependent replication and most HRV strains replicate optimally at 33°C. Delivery of heated, humidified air to the upper airways has the potential to reduce viral replication, but evidence of the effectiveness of this treatment of the common cold is inconclusive. We plan to test the hypothesis that delivery of humidified air heated to 41°C at high flow, nasal high flow rhinothermy (rNHF), for 2 hours daily for five days is more effective in reducing common cold symptom severity and duration than five days of ‘sham’ rhinothermy.
Methods and analysis This is a randomised, single-blind, parallel-group trial comparing rNHF to ‘sham’ rhinothermy in the treatment of common cold. We plan to recruit 170 participants within 48 hours of the onset of symptoms of common cold and randomise them 1:1 to receive one of the two treatments for five days. The study duration is 14 days, which includes clinic visits on the first day of randomisation and four days post-randomisation, and a phone call on the 14th day. Participants will complete daily symptom diaries which include a symptom score, the Modified Jackson Score (MJS). The primary outcome is the MJS after four days.
Ethics and dissemination New Zealand Ethics Registration: 17/STH/174. Results will be published in a peer-reviewed medical journal, presented at academic meetings, and reported to participants.
Trial registration number U1111-1194-4345 and ACTRN12617001340325; Pre-results.
- respiratory infections
- infectious diseases
- internal medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The 35 L/min flow rate using the nasal high flow rhinothermy device will enhance the delivery of heated, humidified air to the upper airways.
Use of ‘sham’ rhinothermy ensures an appropriate control treatment.
Nasopharyngeal samples allow subgroup analysis of subjects which are PCR positive for specific respiratory pathogens.
It is not possible to blind the investigators to the interventions being administered.
Introduction
Common cold
The common cold is the most common infectious disease affecting humans.1 The common cold is a viral upper respiratory tract infection with typical symptoms that include nasal congestion, rhinorrhoea, sneezing, sore throat and cough.2 Many viruses cause the common cold but human rhinoviruses (HRVs) are the most frequent pathogen.3
On average, adults have two to four colds per year.2 The mean duration of symptoms of the common cold is reported to be between seven and 10 days and it is usually a self-limiting disease.2 However, the common cold can cause significant morbidity and has a substantial economic impact on society. In the USA alone, non-influenza viral respiratory tract infections cause an estimated 20 million lost workdays, 110 million physician visits and 6 million emergency department visits annually,4 producing an economic burden of US$40 billion each year.4
Furthermore, HRV infection poses particular risks to those with other comorbidities, such as asthma and chronic obstructive pulmonary disease (COPD), where they are a major cause of disease exacerbations.5 6
No effective treatment has been identified for the common cold and current treatments are mainly symptomatic. There are many cold remedies marketed and, for example, in the USA, an estimated US$2.9 billion are spent on over-the-counter medications for the common cold annually.4 Among the treatments used are zinc, vitamin C, nasal decongestants, non-steroidal anti-inflammatory drugs, echinacea, antihistamines, antibiotics and heated, humidified air. The evidence for the effectiveness of many of these is inconclusive or, at most, demonstrates a limited benefit for symptom reduction.3
Rhinothermy
HRVs cause up to two-thirds of colds, and importantly for the hypothesis tested in this research, have temperature-dependent replication.7 The antiviral effect of temperature on HRVs has been investigated in vitro.8–11 Replication of most HRV strains is optimal between 33°C and 35°C,7 which is the normal physiological temperature of the upper airways.7 Studies report that raising the nasal mucosal temperature may inhibit viral replication8 and this may be a treatment strategy to attenuate the severity and duration of common cold symptoms. Forstall et al 8 report that the greatest inhibition of HRV occurs when exposed to a temperature of 43°C for at least one hour8 and Conti et al report that HRV replication is suppressed when exposed to a temperature of 45°C for 20 minutes.9
Rhinothermy is the delivery of heated, humidified air to the upper airways via the nasal passages. Studies conducted in vivo have delivered heated and humidified air to participants at a range of temperatures, from 40°C to 44°C,8 12–15 and using a variety of devices, including an anaesthetic mask12 and steam-venting nozzles held a distance away from the nares.8 13–15 These delivery systems may be inefficient and inconsistently deliver humidified air to the upper airways at the desired temperatures. A recent Cochrane systematic review of six rhinothermy studies in which participants with the common cold were treated with heated, humidified air did not show any harms or benefits, but advised cautious interpretation of the evidence, concluding that its quality was low grade.1 The review recommended that further double-blind randomised controlled trials (RCTs) are needed, which provide definitive diagnosis derived from viral cultures and use uniform symptom scores.1 It is evident that future trials should use standardised treatment modalities, which effectively and consistently raise the temperature of the upper airways for the duration of treatment.
The technical difficulties in delivering heated, humidified air to the upper airways in an efficient and tolerable manner have been overcome with the nasal high flow rhinothermy (rNHF) device manufactured by Fisher & Paykel Healthcare (Auckland, New Zealand). This device can consistently deliver high-flow, humidified air heated to 41°C, to the upper airways. The device is well tolerated and acceptable to adults.16
A recent feasibility study recruited 30 participants with symptoms of the common cold into an RCT of five days of rNHF therapy or vitamin C with the main patient-reported outcome measure, the Modified Jackson Score (MJS).17 The rNHF therapy delivered 100% humidified air at 35 L/min and 41°C for two hours daily. Compared with vitamin C, rNHF treatment caused a clinically significant five unit reduction in the MJS.
Following this feasibility study, we plan to undertake a definitive, adequately powered RCT to investigate the effectiveness of rNHF treatment in the management of the common cold.
Methods and analysis
Hypothesis
Five days of rNHF therapy is more effective in reducing symptom severity and duration of the common cold than five days of ‘sham’ rhinothermy.
Study design and participants
This is a randomised, single-blind, parallel-group trial that will compare five days of rNHF therapy (100% humidified air at 35 L/min and 41°C for two hours daily) with ‘sham’ rhinothermy therapy (100% humidified air at 10 L/min and 31°C for 10 minutes daily) in the treatment of the common cold.
We plan to recruit 170 participants within 48 hours of the onset of specific symptoms of the common cold. Participants will be randomised 1:1 to receive one of the two possible treatment arms and will receive this treatment for the first five days of the study. Study visits will occur on day one and day five with a follow-up phone call on day 14. The patient-reported outcome measure will be the MJS.18 19
Data collection
All study data will be collected and recorded using the electronic data capture tool REDCap (Research Electronic Data Capture).20 The MJS on day one will be collected by the investigator as part of the screening visit. After this, participants will complete a daily symptom diary, including a daily MJS, remotely, and will enter this data directly into either an electronic case report form (eCRF) on REDCap, or on paper.
Inclusion criteria
Aged 18–75 years.
In the investigator’s opinion, the participant is able and willing to comply with all trial requirements.
Onset of symptoms within the last 48 hours at the time of consent.
MJS of ≥7.
Exclusion criteria
Immunocompromised condition:
Conditions causing immunosuppression, for example, HIV/AIDS and active cancer.
Currently prescribed systemic steroids or other immunosuppressant medication.
Nasal conditions, such as deviated septum and chronic rhinitis, which the investigator considers could impair nasal breathing.
Current use of or requirement for oral antibiotics for respiratory tract infection and pneumonia, or infective exacerbation of underlying respiratory condition.
Current use of or requirement for parenteral antibiotics.
Daily intranasal or inhaled steroids will be allowed if part of the participant’s regular therapy. If not taken prior to enrolment, they should be withheld for the duration of the study.
Have an implantable medical device.
Have a notifiable disease.
Have existing travel plans that require them to leave the greater Wellington region during the first five days of the study, the period during which the participant will be using the rhinothermy device.
A current diagnosis of asthma, COPD or other significant respiratory conditions. (Participants who have not had asthma symptoms nor any requirement for asthma medication within the last 12 months, will be eligible for inclusion in the study.)
A positive GeneXpert point-of-care test for influenza A or B.
The investigator believes that the participant or their caregiver will be unable to safely use rNHF without medical supervision.
Have any other condition that, at the investigator’s discretion, is believed may present a safety risk, or impact the feasibility of the study or the study results.
Study outline
The screening and study visits will follow a standardised schedule of procedures (table 1).
Schedule of procedures
Day one
Screening and study enrolment
The initial screening visit will take place at the Medical Research Institute of New Zealand (MRINZ) study clinic and involves:
Calculation of symptom duration and day one (baseline) MJS.
Assessment for the presence of exclusion criteria.
Obtaining past medical, surgical, smoking, allergy and medication history.
Collection of nasopharyngeal specimens to exclude influenza A or B viruses.
A physical examination to ensure that there are no physical health issues, which may exclude the participant from the study.
Informed consent will be obtained prior to screening (online supplementary file 1: Model screening participant information sheet and consent form) and again prior to enrolment in the study for eligible participants (online supplementary file 2: Model study participant information sheet and consent form). Participants will sign electronic consent forms on REDCap.
Supplementary file 1
Supplementary file 2
Enrolled participants are asked to refrain from using any over-the-counter medication, vitamins or herbal remedies specifically for common cold symptom relief Use of these medications will not constitute a reason for withdrawal but will be documented alongside other collected participant data, either by investigators during study visits, or by the participants themselves in their daily symptom diaries.
Nasopharyngeal specimens
Nasopharyngeal specimens will be obtained during the screening visit using Copan’s flocked swabs (FLOQSwabs). These specimens will be tested for the presence of influenza type A or B virus nucleic acid using the GeneXpert Xpress Influenza/Respiratory Syncytial virus (RSV)point-of-care test (Cepheid, Sunnyvale, California, USA). The sensitivity and specificity of real-time PCR using the GeneXpert Xpress Influenza/RSV point-of-care test for the rapid diagnosis of influenza virus has been reported as 97.8% and 100%, respectively.21
Nasopharyngeal specimens obtained from enrolled participants will also be sent to Canterbury Health Laboratories for multiplex respiratory testing to confirm the presence or absence of 21 different respiratory pathogens: influenza A virus; influenza A (H1N1) pdm 2009 virus; influenza B virus; HRV; human coronavirus NL63, 229E, OC43 and HKU1; human parainfluenza virus 1, 2, 3 and 4; human metapneumoviruses A/B; human bocavirus; human respiratory syncytial viruses A/B; human adenovirus; enterovirus; human parechovirus and Mycoplasma pneumoniae.
Randomisation
Following the screening process, eligible participants who are enrolled in the study will be immediately randomised 1:1 to one of the following treatment arms:
rNHF (n=85 participants).
‘Sham’ rhinothermy (n=85 participants).
A permuted block randomisation method stratified by duration of illness, lesser versus greater than or equal to one day, will be used to allocate participants to either treatment arm. The computer-generated sequence will be supplied by the study statistician, independent of the investigators. The eCRF system will conceal the allocations and will release a participant’s randomisation outcome at the time of randomisation. The randomisation schedule will only be accessed by the study statistician and the eCRF provider; study staff will not have access to the randomisation schedule.
Treatments
There are two randomised treatments:
rNHF: 100% humidified air delivered via the rhinothermy device (Fisher & Paykel Healthcare) at 35 L/min and 41°C for two hours daily for five days.
On day one, this will be administered under the supervision of investigators. On day two to day five inclusive, participants will self-administer the treatment at home. The participant will be encouraged to self-administer their treatment in a single two hour session on each day of treatment. However, if they are unable to do so, then the daily rhinothermy treatment can be split into no more than two sessions aiming for the participant to complete a total of two hours of rhinothermy each day. Participants may also reduce the flow from 35 L/min to 30 L/min at home according to comfort.
‘Sham’ rhinothermy: 100% humidified air delivered via the myAIRVO2 device (Fisher & Paykel Healthcare) at 10 L/min and 31°C for 10 minutes daily for five days.
On day one, this will be administered under the supervision of investigators. On day two to day five inclusive, participants will self-administer the treatment at home. The participant will be encouraged to self-administer the ‘sham’ rhinothermy in a single 10 minute session on each day of treatment.
In contrast to the recent feasibility study, ‘sham’ rhinothermy will be used as the control rather than vitamin C. The use of ‘Sham’ rhinothermy in the control arm will provide the advantage of masking participants to treatment allocation and will control for any placebo effect associated with the use of a rhinothermy device. The rationale for the ‘sham’ device settings is to produce a treatment which is unlikely to have any therapeutic effect. For respiratory pathogens with temperature-dependent replication, a temperature of 31°C should not inhibit viral replication.
General and safety checks will be completed on each device before they are provided for use. Participants will also be asked to read through the relevant device warnings and personally sign and date these to document that they have read and understood them.
Adherence to trial treatment
Both the intervention and control devices automatically record device use data electronically. Adherence to the rNHF and ‘sham’ rhinothermy treatments will be assessed by investigator review of these data. Adherence to rNHF will be defined as a minimum of 90 minutes use per day, delivered in no more than two sessions per day. Adherence to’ sham’ rhinothermy will be defined as a minimum of 6 minutes use per day, delivered in no more than a single session.
Day two to day 14
The day one MJS will be calculated as part of the screening visit and participants will be asked to complete a daily symptom diary online for the subsequent 13 days. The links to the daily symptom diary will be generated by REDCap and sent to the participants via automated email. The daily symptom diary data will be entered directly into REDCap by the participants themselves. Those participants without computer or internet access will be provided with paper versions of the daily symptom diary.
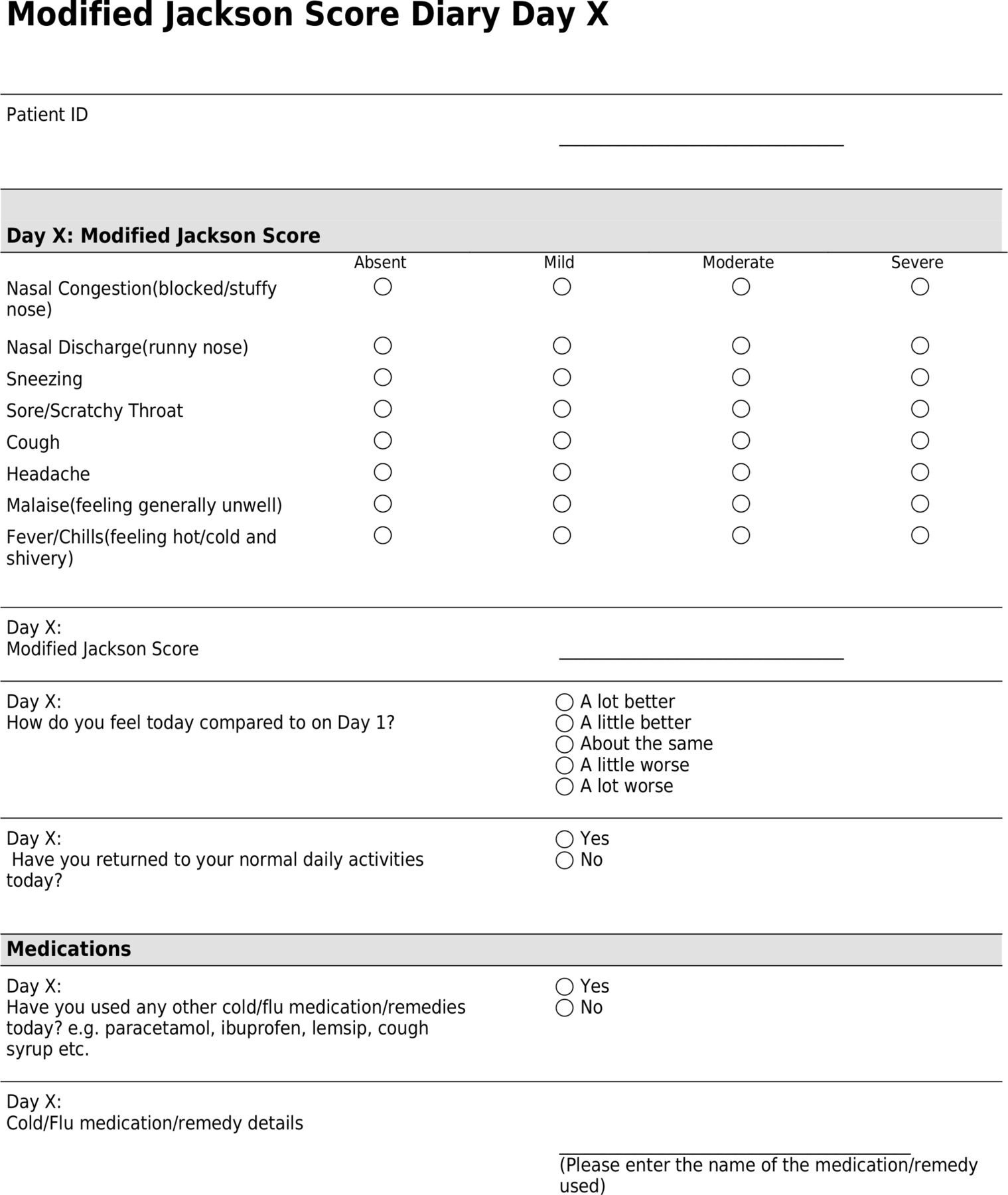
Using an electronic or paper symptom diary (figure 1), participants will document:
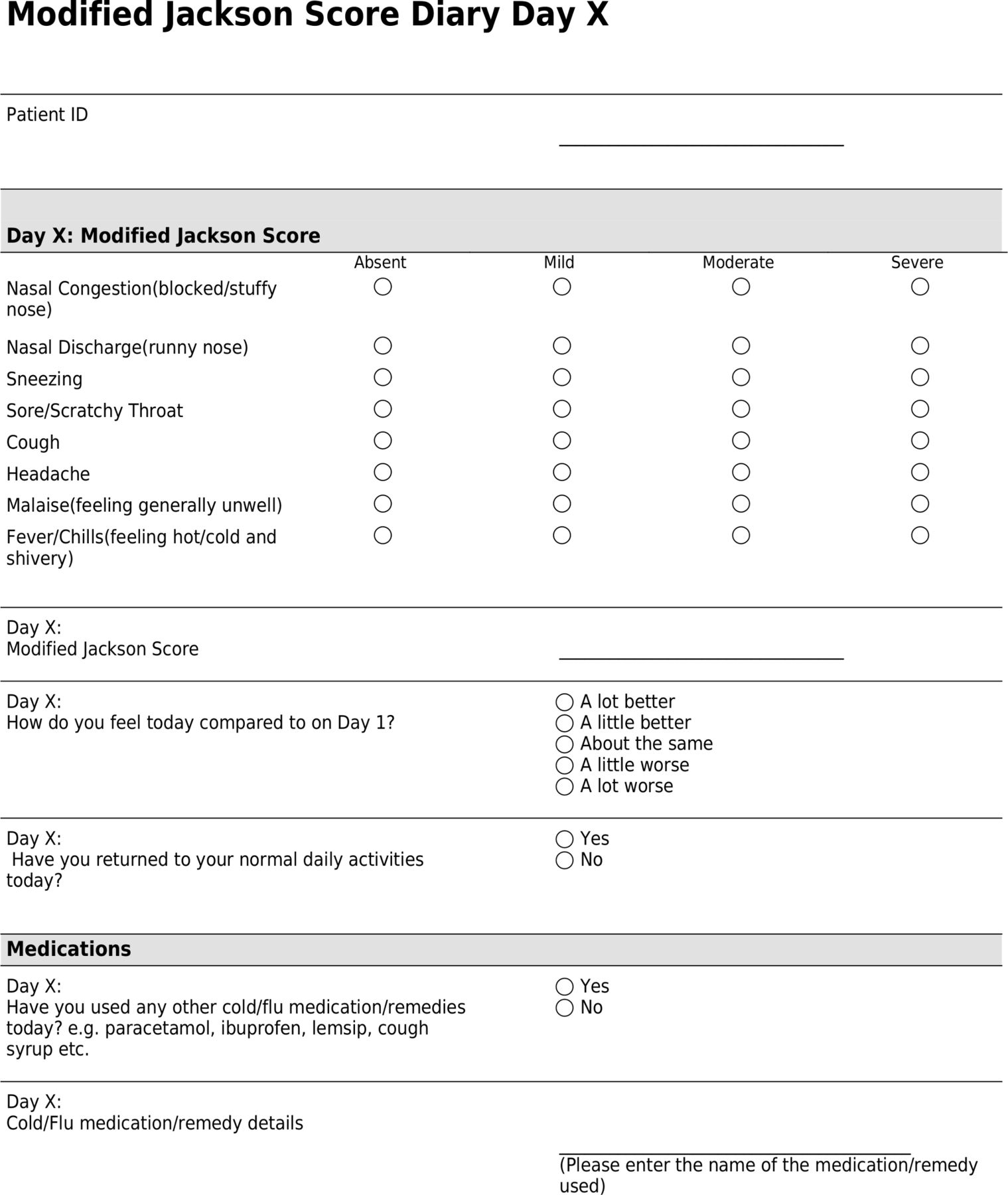
Participant daily symptom diary. ID, identification.
Daily MJS.
How they feel each day, either a lot worse, a little worse, about the same, a little better or a lot better, as compared with day one.
The day during the study when they returned to their usual daily activities.
Use of cold medications/remedies and provide details of these if applicable.
Non-completion of the online symptom diary will trigger a reminder from REDCap.
Day five (study clinic visit)
Participants will return to the MRINZ study clinic for a second visit to return their devices and complete a tolerability questionnaire regarding their device and treatment. Any new medications, adverse events (AEs) or device issues will also be reviewed during this visit.
Participants who have not administered their day five treatment (rNHF/‘sham’ rhinothermy) at the time of attending will be encouraged to receive the allocated treatment during their day five visit.
The investigator will download the device use data from the participant’s device and upload this into RedCap.
Day 14 (follow-up phone call)
Participants will receive a follow-up phone call on the final day of the study (day 14) to review any new medications, AEs or device issues, and confirm the end of the study. Participants will be asked to provide any feedback they consider appropriate, and study reimbursement will be arranged and sent out.
Outcome measures
Primary objective
To estimate the difference in the severity of symptoms of the common cold between rNHF and ‘sham’ rhinothermy after four days, and test the hypothesis that rNHF is superior to ‘sham’ rhinothermy.
Primary outcome measure
Day four MJS (figure 2), captured by participant daily symptom diary.

{kind=link}
{kind=link}
Modified Jackson Symptom Score.
Secondary objectives
To estimate the difference in the severity of symptoms of the common cold over 14 days between five days of rNHF and five days of ‘sham’ rhinothermy.
To estimate the difference in the duration of symptoms of the common cold between five days of rNHF and five days of ‘sham’ rhinothermy.
To estimate the difference in time to feeling ‘a little better’ and ‘a lot better’ compared with day one, between five days of rNHF and five days of ‘sham’ rhinothermy.
To estimate the difference in time until return to ‘normal daily activities’ between five days of rNHF and five days of ‘sham’ rhinothermy.
To estimate if there is an interaction between any treatment effect and HRV viral status.
Secondary outcome measures
MJS on days two, three and five to 14 inclusive.
Time until feeling ‘a little better’ compared with study entry (day one).
Time until feeling ‘a lot better’ compared with study entry (day one).
Time until resolution of symptoms (defined as the start of a 24-hour period in which the MJS was ≤1 and remained so for 24 hours).
Time until return to normal daily activities, as recorded in the daily symptom diary.
MJS on day four, in those participants whose nasopharyngeal specimens are HRV positive.
Tertiary objectives
Description of the viruses affecting participants in the study.
To determine the tolerability of rNHF therapy.
To determine patterns of use of the rNHF device over the treatment period, including the number of days used and hours of use per day.
To determine the adherence to rNHF therapy.
Tertiary outcome measures
Proportion of isolates from each pathogen type identified by real-time PCR analysis of nasopharyngeal specimens obtained on day one, for example, rhinovirus, coronavirus, respiratory syncytial virus, and so on.
Tolerability questionnaire results.
Device use data downloaded from the rNHF device.
Statistical analysis
Statistical analyses will use the intention-to-treat principle and all participants who are randomised will be included in the analysis. The primary comparison is between rNHF treatment and ‘sham’ rhinothermy treatment. The primary outcome variable will be analysed by analysis of covariance (ANCOVA) with the day one (baseline) MJS as the covariate. An interaction analysis will be used to assess evidence that there are different treatment outcomes for those participants who test positive for HRV. A sensitivity analysis will be used to assess evidence that there is an interaction between the adherence and the primary outcome. The type I error rate for the primary analysis will be at a p value of 0.05.
Any deviation(s) from the original statistical plan will be described and justified in the protocol or final report, as appropriate.
Sample size and power calculation
In our recent RCT of rNHF compared with vitamin C, a 5-unit reduction in MJS represented a substantial clinical benefit.16 The upper confidence limit for the SD in this trial was 6.6. A sample size of 76 in each group allows the detection of a difference of 3.5 units, with 90% power and a type I error rate of 5%. This feasibility study had a dropout rate of zero; however, even though the likelihood of dropouts in our study is low, in order to ensure that this study remains adequately powered, we will allow for a 10% dropout rate in our power calculation. Allowing for a 10% dropout rate, a total of 85 participants will be randomised to each group.
There will not be an interim analysis.
Ethics and dissemination
Locality approval has been obtained from all recruitment sites (MRINZ and Capital & Coast District Health Board). Medsafe has been informed in writing of our intentions to undertake this study.
Patient and public involvement
Participants have not been directly involved in the development of the research questions, outcome measures or design of this study. This study protocol has been developed based on previous similar studies and the existing medical literature. As outlined in the introduction, review of the literature indicates that it is in the public interest to conduct research that investigates treatments for the common cold. Reduction in symptom severity and duration is a priority for individuals with the common cold. As such, this study includes outcome measures that will investigate the effect of rhinothermy on symptom severity and duration of illness.
A recent feasibility study that involved 30 participants has provided information related to effective recruitment methods, the tolerability of the device and adherence. This feasibility study has shown that the rhinothermy device is well tolerated16; however, to further consolidate these findings and assess treatment burden, this study will include outcome measures those look at adherence to and tolerability of the treatment. Participants will also be encouraged to provide feedback or raise any concerns with regards to any aspect of the study.
Once completed, the results of the study will be submitted for publication. The results will also be disseminated to those participants who indicated that they wished to receive them.
Reimbursement
Participants will be eligible for monetary reimbursement for completing the study. A standard agreed amount has been decided, which will cover any expenses associated with participation in the study. Participants will be eligible for reimbursement on completion of the study and this amount will be either the full agreed amount for completing the study in its entirety, or a reduced amount proportional to their involvement in the study.
Protocol amendments
The principal investigator (PI) will submit all changes to relevant parties (including the study funder) and all substantial amendments to the original approved documents will be submitted to the Health and Disability Ethics Committee for ethical review. Study recruitment will be paused until any substantial amendments have been approved by the ethics committee. Protocol changes will also be submitted to the Australian New Zealand Clinical Trials Registry.
Participant safety
The investigator will ensure that this trial is conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice.
AEs and device deficiencies
As sponsor, Fisher & Paykel Healthcare will monitor the study according to their own procedures and will review all serious AEs (SAEs). All AEs and device deficiencies will be reported to the study sponsor in accordance with current local and national adverse event reporting policies. The sponsor will manage device deficiency reports as part of its ongoing post-marketing safety review process.
The sponsor will report all AEs and relevant investigational device deficiencies to the regulatory authorities and PI within the required time period, in accordance with current reporting requirements.
Data Safety Monitoring Committee
A Data Safety Monitoring Committee (DSMC) will be appointed to review all SAEs on an expedited basis. To ensure adequate study safety, they will review enrolments, withdrawals and AEs every 6 months. The DSMC may recommend to the sponsor that the study should be terminated; however, the final decision will be made by the sponsor.
Dissemination
The PI and the corresponding author will have full access to the study data and final responsibility for submitting the report for publication. The PI will also take responsibility for reporting the results to those participants who have expressed that they would like to receive the study results. Publication of the study outcomes will comprise publication of the study as a whole and is encouraged by the sponsor regardless of the outcome. The sponsor will have no involvement in collection, analysis and interpretation of data; preparation of the report or decision to submit for publication.
References
Footnotes
Contributors GB wrote the current HDEC approved version of this protocol and the protocol manuscript with input from IB and JF. RB, IB, JF and AS designed the study and directed the project. MW designed and verified the randomisation schedule and statistical analysis methods. MD and LJ advised on collection and laboratory analysis of nasopharyngeal specimens. SM was an investigator in the rhinothermy in the common cold feasibility study and as such contributed to the development of this protocol. JH and IK contributed to protocol amendments.
Funding The study is funded by the Fisher & Paykel Healthcare (FPH). FPH is also providing the rhinothermy and myAIRVO2 (‘sham’ rhinothermy) devices and associated consumables.
Competing interests None declared.
Ethics approval The protocol has been approved by the Health and Disability Ethics Committees of New Zealand and the Regional Advisory Group—Māori (RAG-M).
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.