Article Text

Abstract
Introduction Ventilator-associated pneumonia (VAP) is the most common healthcare-associated infection in critically ill patients. Prior studies suggest that probiotics may reduce VAP and other infections in critically ill patients; however, most previous randomised trials were small, single centre studies. The Probiotics: Prevention of Severe Pneumonia and Endotracheal Colonization Trial (PROSPECT) aims to determine the impact of the probiotic Lactobacillus rhamnosus GG on VAP and other clinically important outcomes in critically ill adults.
Methods PROSPECT is a multicentre, concealed, randomised, stratified, blinded, controlled trial in patients ≥18 years old, anticipated to be mechanically ventilated ≥72 hours, in intensive care units (ICUs) in Canada, the USA and Saudi Arabia. Patients receive either 1×1010 colony forming units of L. rhamnosus GG twice daily or an identical appearing placebo. Those at increased risk of probiotic infection are excluded. The primary outcome is VAP. Secondary outcomes are other ICU-acquired infections including Clostridioides difficile infection, diarrhoea (including antibiotic-associated diarrhoea), antimicrobial use, ICU and hospital length of stay and mortality. The planned sample size of 2650 patients is based on an estimated 15% VAP rate and will provide 80% power to detect a 25% relative risk reduction.
Ethics and dissemination This protocol and statistical analysis plan outlines the methodology, primary and secondary analyses, sensitivity analyses and subgroup analyses. PROSPECT is approved by Health Canada (#9427-M1133-45C), the research ethics boards of all participating hospitals and Public Health Ontario. Results will be disseminated via academic channels (peer reviewed journal publications, professional healthcare fora including international conferences) and conventional and social media. The results of PROSPECT will inform practice guidelines worldwide.
Trialregistration number NCT02462590; Pre-results.
- critically ill
- infection
- intensive care
- probiotics
- ventilator-associated pneumonia
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Randomized placebo controlled multicentre trial.
Evaluation of the effect of probiotics on pneumonia, other intensive care unit (ICU)-acquired infections and diarrhoea in a large, adequately powered trial.
International enrolment including patients over 65 years of age to enhance the generalizability of the findings.
Characterisation of pre-hospital frailty to help understand the relationship between frailty, probiotics and ICU-acquired infections.
Severely immunocompromised patients are excluded for safety reasons.
Introduction
Ventilator-associated pneumonia (VAP) is the most common healthcare-associated infection in critically ill patients, and is associated with a significant burden of disease.1 In a systematic review, the pooled incidence of VAP in patients mechanically ventilated for >48 hours ranged from 10%–23%, and VAP conferred a twofold attributable-risk of dying in the intensive care unit (ICU), with an attributable cost ranging from USD$10 000–$13 000 per patient.1 Therefore, preventing VAP is a patient safety priority.2 3
Unfortunately, VAP prevention strategies are variably applied in practice,4 which underscores the need for simple, safe, effective and affordable VAP reduction strategies. Probiotics may represent one such novel approach. Probiotics have emerged as a biologically plausible strategy to prevent VAP, through influencing microbiota, enhancing gut barrier function and reducing pathogenic bacterial load.5–8 Systematic reviews suggest that probiotics reduce VAP by 25%–30% when compared with placebo.9–11 However, most previous randomised trials were small, single centre studies. Meta-analyses of small single centre trials often yield implausibly large treatment effects.12 13 Hence, the clinical benefits of probiotics may be overestimated, and a large, well-powered multicentre trial is needed.
In a recent trial sequential meta-analysis of randomised trials testing the effect of probiotics on VAP during critical illness, 11 of 13 included trials evaluated a Lactobacillus species alone or in combination, and 2 of these trials used Lactobacillus rhamnosus GG,14 including the most rigorous trial by Morrow et al.15 This high quality trial compared L. rhamnosus GG to corresponding placebos in 146 patients and the patients treated with L. rhamnosus GG had lower rates of VAP suggesting that L. rhamnosus GG, specifically, is a promising probiotic to prevent VAP in a selected high-risk ICU population.15
We recently completed the Probiotics: Prevention of Severe Pneumonia and Endotracheal Colonization Trial (PROSPECT) pilot (www.clinicaltrials.gov NCT01782755)16 in 14 ICUs which compared L. rhamnosus GG to placebo in critically ill mechanically ventilated patients. The feasibility objectives of the pilot trial were related to (1) recruitment: at least two patients per ICU per month; (2) maximal protocol adherence: ≥90% of prescribed doses are actually administered; (3) minimal contamination: <5% of patients receive a single dose of open-label probiotics and (4) outcome incidence: at least 10% of enrolled patients developed VAP. The pilot trial met all four feasibility outcomes: (1) 150 patients were enrolled over 11 months, with 1.9 patients per ICU per month; (2) adherence to study product was 97.4%; only 2.6% of doses prescribed were not received; (3) contamination did not occur; no patients received a dose of open-label probiotic at any time; and (4) the adjudicated VAP rate was 19%.17 Therefore we launched PROSPECT—a multicentre randomised concealed stratified blinded parallel-group placebo-controlled superiority trial to determine whether the probiotic L. rhamnosus GG compared with placebo reduces VAP and other clinically important outcomes in critically ill mechanically ventilated patients (www.clinicaltrials.gov NCT02462590). In this paper, we summarise the protocol (research ethics board (REB)-approved version, version 1.0, date: 27 February 2015) and statistical analysis plan (version 2.0, date 17 May 2018) for PROSPECT’s primary analysis, reported using both the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines which define standard protocol items for clinical trials18 and recent statistical analysis plan guidelines.19
Methods
Trial population and eligibility
Patients will be recruited from 44 ICUs in Canada, the USA and Saudi Arabia (detailed list of study sites available (www.clinicaltrials.gov NCT02462590)). The inclusion and exclusion criteria are presented in box 1. Following completion of the PROSPECT pilot,16 17 the exclusion criteria were refined, informed by an extensive literature review focused on the safety or harm of Lactobacillus spp. probiotic administration,20 experience with probiotics in the pilot trial,17 and following discussions with the PROSPECT Steering Committee and the Canadian Critical Care Trials Group21 (box 1 footnote for details of changes).
Inclusion and exclusion criteria
Inclusion criteria
Adults ≥18 years of age admitted to a medical, surgical or trauma ICU.
Receiving invasive mechanical ventilation, estimated to be required for ≥72 hours.
Exclusion criteria
Invasively mechanically ventilated >72 hours at the time of screening.
Potential increased risk of iatrogenic probiotic infection including specific immunocompromised groups: HIV <200 CD4 cells/µL, chronic immunosuppressive medications, previous transplantation at any time, chemotherapy in the last 3 months, absolute neutrophil count <500. Previous or current corticosteroids use is not exclusionary.
Risk for endovascular infection: rheumatic heart disease, congenital valve disease, surgically repaired congenital heart disease, unrepaired cyanotic congenital heart disease, valvular replacement (mechanical or bio-prosthetic), previous or current endocarditis, permanent endovascular devices (eg, endovascular grafts, inferior vena cava filters, dialysis vascular grafts), tunnelled hemodialysis catheters, pacemakers or defibrillators. These are not exclusions: coronary artery stents or bypass grafts, mitral valve prolapse, bicuspid aortic valve, temporary catheters (central venous, peripherally inserted, extra-corporeal life support-related) or neurovascular coils.
Primary diagnosis of severe acute pancreatitis.
Percutaneously inserted feeding tubes in situ, as per Health Canada.
Strict contraindications or inability to receive enteral medications.
Intent to withdraw advanced life support.
Previous enrolment in this trial or current enrolment in a potentially confounding trial.
*Changes from the Probiotics: Prevention of Severe Pneumonia and Endotracheal Colonization Trial pilot are as follows: 1. Omitted radiation therapy as an exclusion criterion; 2. Omitted steroid exposure as an exclusion criterion; 3. Better defined transplant to explicitly exclude all transplant patients (autologous stem cell patients are now excluded); 4. Better defined the cardiac valvular diseases at risk; 5. Removed surgery of oesophagus/stomach/small bowel as exclusion criteria and replaced with any strict contraindication or inability to receive enteral medications; 6. Replaced severe acute pancreatitis with organ dysfunction with primary diagnosis of severe acute pancreatitis; 7. Omitted pregnancy as exclusion criterion.20
Consent and randomisation
Research Coordinators screen all mechanically ventilated patients for potential trial enrolment, recording those that meet individual inclusion and exclusion criteria. Once eligibility is confirmed, a priori written informed consent or deferred consent is obtained from the patient or substitute decision maker as per our consent guidelines,22 23 and according to local ethics approval. The patients are allocated to treatment in a 1:1 ratio via a computer-based random number generator in variable unspecified block sizes, stratified by centre and by medical, surgical or trauma admission status.
Blinding
Patients, bedside clinicians, investigators and research coordinators are blinded to allocation. Study pharmacists at each centre are not blinded; they randomise patients and prepare study product for administration without being involved in the day-to-day bedside care of patients. The biostatisticians will remain blinded until the main analysis is complete. Unblinding will not be permissible throughout the trial.
Interventions and comparator
Patients in the intervention group receive 1×1010 colony forming units of L. rhamnosus GG (i-Health, Inc.) in one capsule suspended in tap water or sterile water (dependant on local practices), administered through a nasogastric or orogastric feeding tube. Patients in the placebo group receive an identical capsule containing microcrystalline cellulose. The same dose of microcrystalline cellulose is present in the L. rhamnosus GG capsules. Patients receive study product post randomisation until: (1) ICU discharge or death; or (2) 60 days in the ICU; or (3) isolation of Lactobacillus spp. in a culture from a sterile site or if it is the sole or predominant organism in a culture from a non-sterile site.
The intervention is packaged in blister-cards of 10 capsules. For quality assurance purposes, we are performing an independent quality assessment of the study product supplied throughout the trial.24 One randomly selected capsule from every 10th card of both probiotic and placebo is cultured in the Surette Microbiome Laboratory at McMaster University (Hamilton, Ontario), to ensure the dose and integrity of both the study product and placebo, as successfully done in the pilot trial.16 17
Data collection
Research Coordinators collect data at baseline (eg, demographics, illness severity, life support using the Acute Physiology and Chronic Health Evaluation (APACHE) II score), and daily (eg, study product administration, VAP prevention strategies and other cointerventions), and all primary and secondary outcomes (online supplementary appendix 1) by completing data collection forms22 and uploading to a secure web-based electronic data capture system (iDataFax, Seattle, Washington). To protect the personal health information of patients enrolled, all identifying information will be de-linked. Participants will be assigned a unique identification code (study ID). The code-breaking information will be kept separate from the data extraction files. It will be the responsibility of the site investigators to ensure that the code-breaking information is totally inaccessible to individuals who are not on the research team. Personal health information about enrolled participants will include age, sex and admitting diagnosis, but will be de-identified at the recruiting centre and anonymised in the main database over the course of the trial and thereafter.
Supplemental material
Outcomes
Primary outcome
The primary outcome is adjudicated VAP. Clinically suspected VAP at participating sites is being centrally adjudicated independently and in duplicate by two physicians blinded to allocation and centre, informed by the following standardised definition: receiving invasive mechanical ventilation for >2 days, when there is a new, progressive or persistent radiographic infiltrate on chest radiograph plus any 2 of the following: (1) fever (temperature >38°C) or hypothermia (temperature <36°C); (2) relative leucopenia (<3.0×106/L) or leucocytosis (>10×106/L) and (3) purulent sputum.25 As the American College of Chest Physicians (ACCP) definition did not provide thresholds for leucopenia or leucocytosis, the thresholds were obtained from Morrow et al 15 as their VAP definition was also based on the ACCP definition.25 Any disagreement in adjudication will be resolved through discussion and consensus. Acknowledging that there is no universally accepted gold standard VAP definition,26 and that in non-immunocompromised patients, routine invasive testing is not associated with improved outcomes,27 we are also collecting data to allow VAP reporting according to several other definitions.28–31
Secondary outcomes
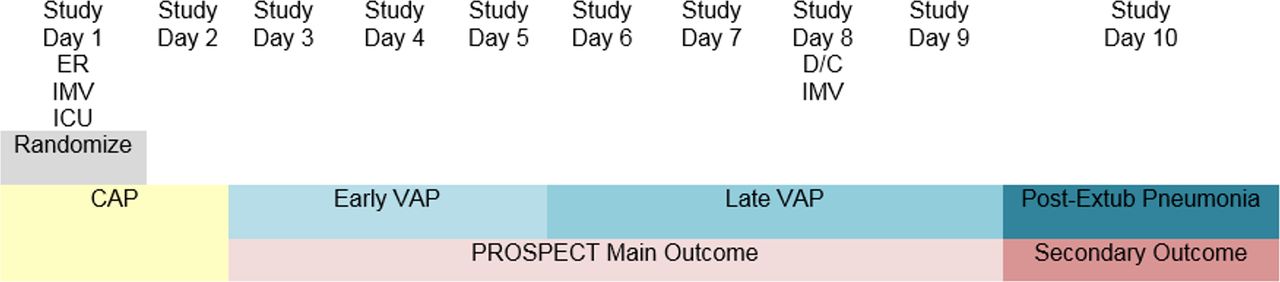
Early VAP, late VAP and post-extubation pneumonia: We are classifying VAP by early VAP and late VAP, as the aetiological organisms may differ, the antimicrobials prescribed may differ and the prognosis is often worse for late VAP.32 33 Early VAP is defined as pneumonia arising on day 3, 4 or five after the initiation of mechanical ventilation. Late VAP is defined as VAP arising on day 6 of mechanical ventilation or later, and including up to 2 days after discontinuation of mechanical ventilation (also relevant for patients with a tracheostomy). We are also recording pneumonia arising in the ICU following discontinuation of mechanical ventilation (three or more days after discontinuation), labelled post-extubation pneumonia, to avoid suppressing potentially relevant lung infections that arise in ICU (figures 1 and 2). We will also report a composite outcome of early VAP, late VAP and post-extubation pneumonia, adjudicated independently and in duplicate by two physicians. For the timing of all pneumonia outcomes, we use days rather than hours to inform the classification.
Clostridioides difficile in the ICU and prior to discharge from hospital: diarrhoea (as defined in (d)) and laboratory confirmation of C. difficile or colonoscopic or histopathological findings demonstrating pseudomembranous colitis,34 which will also be adjudicated independently and in duplicate by two physicians.
Any infection acquired during the ICU stay, including bloodstream infection, intravascular catheter-related bloodstream infection, intra-abdominal infection, C. difficile infection, urinary tract infection, skin and soft tissue infection, and others. These individual infections are classified using definitions adapted from the International Sepsis Forum Consensus Conference on Definitions of Infection in the ICU,29 as adapted in prior studies.28 We will also report a composite outcome of any infections (including pneumonia) acquired during the ICU stay. Secondary infectious outcomes (other than pneumonia and C. difficile) are being centrally adjudicated by one physician blinded to allocation and centre, based on review of data collected at each participating site.
Diarrhoea in the ICU: We will record each bowel movement and define diarrhoea incorporating two metrics; the WHO definition (≥3 loose or watery bowel movements per day35), and the Bristol Stool classification for loose or watery stool (type 6 or 7).36
Antibiotic-associated diarrhoea in the ICU: diarrhoea (as defined above in (d)) following the administration of antibiotics, any day antibiotics are administered or within 1 day after starting any antibiotic.37
Antimicrobial use in ICU: defined as daily doses of therapy, defined daily dose and antimicrobial-free days.38 39 Only systemic antimicrobials will be captured (eg, parenteral, intravenous, oral, enteral) whether prophylactic or therapeutic in intent. Topical creams, eye/ear drops and inhaled antimicrobials will be excluded.
Duration: mechanical ventilation, ICU stay and hospital stay.
ICU mortality and in-hospital mortality.
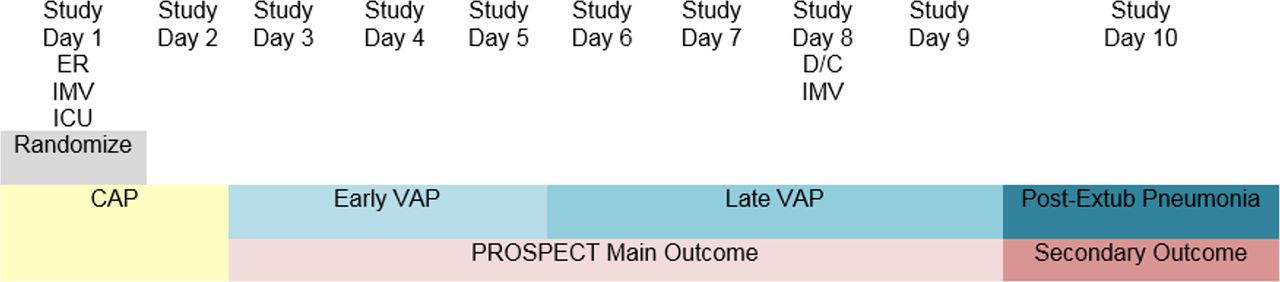
Pneumonia classifications that could arise in patients who require ICU admission and invasive mechanical ventilation at the time of presentation to the emergency room, and are randomised into PROSPECT that day. The primary outcome is adjudicated VAP (any, including early or late), arising on study day 3 or later. Secondary outcomes illustrated include early VAP, late VAP and post-extubation pneumonia. These figures illustrate the pneumonia classification that we are using, according to when the lung infection develops in a patient’s hospital trajectory. The different classifications over time in each example relate to the day of hospital admission, day of ICU admission, day of initiation of mechanical ventilation (via endotracheal intubation or tracheostomy), day of randomisation in the trial and day of discontinuation of mechanical ventilation. Note that the pneumonia classifications over time do not reflect persistent or progressive lung infections, but rather the pneumonia classification that would be ascribed if a new infection develops on each day shown. CAP, community-acquired pneumonia; ER, emergency room; Extub, Extubation; HAP, hospital-acquired pneumonia; ICU, intensive care unit; ICUAP, intensive care unit associated pneumonia; IMV, invasive mechanical ventilation; PROSPECT, Probiotics: Prevention of Severe Pneumonia and Endotracheal Colonization Trial; Rand, randomisation; VAP, ventilator-associated pneumonia.

{kind=link}
{kind=link}
Pneumonia classifications that could arise in patients who require ICU admission and invasive mechanical ventilation after an initial hospital stay, and are randomised into PROSPECT the day following ICU admission. The primary outcome is adjudicated VAP (any, including early or late), arising on study day 3 or later. These figures illustrate the pneumonia classification that we are using, according to when the lung infection develops in a patient’s hospital trajectory. The different classifications over time in each example relate to the day of hospital admission, day of ICU admission, day of initiation of mechanical ventilation (via endotracheal intubation or tracheostomy), day of randomisation in the trial and day of discontinuation of mechanical ventilation. Note that the pneumonia classifications over time do not reflect persistent or progressive lung infections, but rather the pneumonia classification that would be ascribed if a new infection develops on each day shown. CAP, community-acquired pneumonia; ER, emergency room; HAP, hospital-acquired pneumonia; ICU, intensive care unit; IMV, invasive mechanical ventilation; PROSPECT, Probiotics: Prevention of Severe Pneumonia and Endotracheal Colonization Trial; Rand, randomisation; VAP, ventilator-associated pneumonia.
Serious adverse events
In PROSPECT, a seriously adverse event (SAE) is defined as isolation of Lactobacillus spp. in a culture from a sterile site or as the sole or predominant organism cultured from a non-sterile site and results in: (1) persistent or significant disability or incapacity; (2) that is life-threatening or (3) that results in death.40 The rationale for our approach to SAEs accords with our guidelines for academic drug trials in critical care.41 Any culture obtained by the ICU team and processed by the clinical microbiology laboratory as positive for Lactobacillus spp. is recorded. Any such bacterial sample is sent to a McMaster University research laboratory for strain genotyping to evaluate consistency with the administered L. rhamnosus GG strain.
Sample size and power
Based on an estimated 15% VAP rate, 2650 patients will be required to detect a 25% relative risk reduction (RRR) (and absolute risk reduction of 3.75%) with 80% power (alpha 0.05) (table 1). The estimated 15% VAP rate is based on the PROSPECT pilot (adjudicated VAP rate of 19%17) and the REDOXS trial (14%28). The 25% RRR was observed in our meta-analysis of probiotics versus placebo9 and a 24% RRR was found in a recent meta-analysis11 and is more conservative than the 30% RRR in a Cochrane analysis.10 Thus, we will enrol 1325 patients/group (2650 patients). Based on our pilot trial recruitment, we anticipate enrolling approximately 1.9 patients/month/site.17
Determination of the sample size: based on an estimated 15% ventilator-associated pneumonia rate, 2650 patients (n=1325 in each arm) will be required to detect a 25% relative risk reduction with 80% power
Central statistical monitoring
Thrice yearly throughout the trial, we will perform central statistical monitoring by analysing site-specific data receipt and completeness, to help identify and overcome barriers to timely data completion. We will also monitor the proportion of non-screening weeks, and number and reasons for eligible non-randomised patients, to identify and remediate potential recruitment challenges.
We will monitor and report other types of protocol adherence.42 We will track categories such as admissible protocol deviations for clinically justified reasons (eg, strict nil per os status for possible bowel perforation) and logistical reasons (eg, patient discharged early from the ICU so no evening dose given) as distinct from oversights which are protocol violations (eg, dispensing errors). Thus, our protocol adherence regarding non-receipt of study product allows for sensible bedside decision-making, according to metrics from our prespecified taxonomy.43
Statistical analysis
Patients randomised in PROSPECT will be analysed according to the intention-to-treat principle for the main analysis. We will present baseline characteristics of the two groups, including demographic and life support characteristics, and all prevalent infections. Infections will be defined as prevalent if present the day of, or diagnosed 1 day after randomisation (the latter presumed to have started the day of randomisation). For example, prevalent pneumonia could include any patient with pneumonia (community-acquired, healthcare-associated or ventilator-associated) present the day of or the day after randomisation; this classification of pneumonia as prevalent relates only to timing of randomisation and is independent of timing of intubation. Prevalent infections will not be considered outcomes for the trial because they are present at the time of randomisation and are not plausibly modified by probiotics. All prevalent infections will also be centrally adjudicated by one physician blinded to allocation and centre, based on review of data collected at each participating site. A Consolidated Standards of Reporting Trials flow diagram will be generated, representing all randomised patients, their outcomes, the number and reasons for any consent withdrawals or loss to follow-up, as well as eligible non-randomised patients.44
The main analysis will be a Cox proportional hazards analysis evaluating the primary outcome of VAP. This time-to-event analysis will use all information up to the time of censoring such that patients remain in the denominator and contribute information while they are at risk. The assumption for this analysis is that censoring is uninformative. The Cox model will be stratified by: (a) centre and (b) medical versus surgical versus trauma admission diagnosis, reflecting the stratification variables for randomisation. The only independent variable will be randomised treatment group. We will present Kaplan-Meier curves for the primary outcome. We will also report VAP incidence rate, as number of VAP cases per 1000 ventilator days.45 We will report exposures during the ICU stay as is customary for critical care trials (eg, advanced life supports) and cointerventions (eg, pneumonia prevention strategies) relevant for this research question.
For the dichotomous secondary outcomes, we will also use time-to-event analyses. HRs and associated 95% CIs will be estimated using a stratified Cox proportional hazards model.
For continuous outcomes, we will report estimates of the difference, 95% CIs and associated p values. For the continuous outcomes which are often skewed (eg, duration of ventilation, ICU stay and hospital stay), we will first log-transform these variables to see if they become normally distributed; if so, we will use parametric methods on the log-transformed variables to compare between groups. If not, we will compare the two groups using a non-parametric approach on the non-transformed variables. All secondary analyses will be adjusted for the stratification variables used at randomisation (ie, centre and admission diagnostic category (medical, surgical, trauma)).
For the main analysis, when there is a statistically significant difference in binary outcomes, we will calculate other metrics. For example, depending on the results, these may be expressed as the number needed to prophylax with probiotics to prevent 1 case of pneumonia, or the number needed to harm to cause one case of iatrogenic infection with L. rhamnosus GG.
We do not anticipate missing any covariates for the primary outcome analysis—the only independent variable is treatment versus control, and the stratification variables are captured in the randomisation system. We anticipate very little missing outcome data, since most data are collected in the ICU (except hospital vital status and length of stay, and C. difficile infection which is also recorded following ICU discharge in the hospital). For any other outcome that is missing for more than 2% of the patients, we will perform multiple imputation analysis.46–48
We will use graphics and other relevant methods to examine the residuals to assess model assumptions and goodness-of-fit including the proportional hazards assumption for Cox regression analyses.49–51
All estimates of effect will be reported to two decimal places. P values will be reported to three decimal places with those less than 0.001 reported as p<0.001. The criterion for statistical significance will be set at alpha=0.05, using 2-sided tests, but adjusted appropriately for the two planned interim analyses (baseline characteristics, primary and secondary outcomes, adverse events and SAEs) using the Peto-Haybittle approach52 53; the interim analyses will occur at one third and two thirds of total enrolment, performed when complete ICU data are available for 883 and 1766 patients, respectively. Two-sided tests will be used, with a fixed conservative α=0.001 for the first and second interim analyses, and α=0.05 for the final analysis.52 53 Secondary and subgroup analyses will not be adjusted for multiple analyses since these are exploratory.54 All analyses will be performed using the most up-to-date version of SAS.
Following the publication of PROSPECT, the dataset will be used to design observational studies addressing additional hypothesis-driven questions (eg, predictors of diarrhoea and ICU-acquired C. difficile). Access by other PROSPECT investigators will follow a submitted rationale, analysis plan and approval by relevant REBs in accordance with data sharing policies extant at the time of the request.
Sensitivity analyses
We will conduct four sensitivity analyses. To the extent that these sensitivity analyses yield similar results to the main analysis, inferences about the primary outcome will be strengthened.55 56
In case the exact timing of the onset of VAP is uncertain, we will compare the proportion of patients with VAP in the two groups using the Mantel-Haenszel Chi square test, stratified by centre and medical versus surgical versus trauma. Thus, in this sensitivity analysis we will not use a time-to-event approach.
We will check for competing risks to address the problem that those who die can no longer develop VAP. We will analyse PROSPECT to explicitly account for death as a competing risk using the Fine and Grey proportional sub-distribution hazards model.57 58 This analysis will not assume that the censoring of deaths is uninformative; rather, it will assume that deaths could be informative. The rationale for this sensitivity analysis is to assess the robustness of the main findings.55
We will conduct an efficacy analysis of each incident infection, and a composite of all incident infections, restricted to patients who received study product on ≥90% of study days. The rationale for this sensitivity analysis is to investigate the effect of probiotics under conditions of maximal exposure.59
We will include all VAP events that occur after the day of randomisation. The rationale for this sensitivity analysis is that pneumonia arising the day after randomisation may be less likely to be influenced by study product exposure than pneumonia arising two or more days after initial study product exposure.
Subgroup analyses
We will conduct five subgroup analyses based on baseline characteristics. These will evaluate whether these five baseline characteristics have an ‘effect modification’ when the effect of probiotics versus placebo on VAP is compared.60 61 Subgroup analyses will only be performed for the primary outcome.
We will conduct subgroup analyses among medical versus surgical versus trauma patients (the latter defined as patients cared for by a trauma service). We hypothesise that in medical patients, the treatment effect may be attenuated due to more risk factors for VAP that are non-modifiable when compared with surgical or trauma patients.1 To perform this subgroup analysis, we will run the primary Cox regression analysis except that we will include medical versus surgical versus trauma as an independent variable instead of stratifying by it. We will also include the interaction term between medical versus surgical versus trauma and randomised treatment.
We will conduct subgroup analyses based on age (>75 years of age versus 65–75 years versus <65 years). Although little is known about the effects of probiotics in the elderly,62 63 we hypothesise that if, overall, probiotics are associated with a lower rate of VAP than placebo, the treatment effect will be attenuated among older patients because immunosenescence renders their risk of infection less modifiable than younger patients. To perform this subgroup analysis, we will add age >75 vs 65–75 years vs <65 years as an independent variable as well as its interaction with randomised treatment group to the primary Cox regression model.
We will conduct subgroup analyses of the effect of probiotics on VAP after accounting for frailty, defined as a baseline Clinical Frailty Score of >5 out of 9.64 We hypothesise that if, overall, probiotics are associated with a lower rate of VAP than placebo, the treatment effect will be attenuated among patients who are frail, as their risk of infection may not be modifiable. To perform this subgroup analysis, we will add baseline Clinical Frailty Score of >5 as an independent variable as well as its interaction with randomised treatment group to the primary Cox regression model. We began measuring frailty in response to a Canadian research mandate,65 and did not start documenting frailty until 483 patients were enrolled. Thus, rather than imputing frailty status, we will restrict this subgroup analysis to patients enrolled thereafter.
We will conduct subgroup analyses among patients who received antibiotics for 2 days prior to randomisation and the day of randomisation versus patients who did not receive antibiotics for 2 days prior to, or the day of, randomisation. We hypothesise that if, overall, probiotics are associated with a lower rate of VAP than placebo, the treatment effect will be attenuated in patients without recent antibiotic exposure when compared with patients with antibiotic exposure. To perform this subgroup analysis, we will add antibiotic exposure prior to randomisation defined as those receiving antibiotics for 2 days prior to randomisation and the day of randomisation as an independent variable as well as its interaction with randomised treatment group to the primary Cox regression model.
We will conduct subgroup analyses on patients with prevalent pneumonia versus no prevalent pneumonia. We hypothesise that if overall, probiotics are associated with a lower rate of VAP than placebo, the treatment effect will be attenuated among patients with pre-randomisation pneumonia due to challenges interpreting whether the prevalent pneumonia has resolved prior to the development of another pneumonia event. To perform this subgroup analysis, we will add prevalent pneumonia as an independent variable as well as its interaction with randomised treatment group to the primary Cox regression model.
Steering committee
The PROSPECT Steering Committee is responsible for overseeing the conduct of the trial, for upholding or modifying study procedures as needed, and addressing any challenges with protocol implementation. They advise as necessary on operational issues arising that are clinical, methodological, biostatistical or ethical. The steering committee will review any proposed protocol amendment prior to dissemination of the revised protocol to participating centres by email correspondence, and shared on conference calls or webinars. As enrolment ensues, they share new emerging clinical, laboratory or epidemiology information that may impact on the trial. The Steering Committee has discussed and approved the interim statistical analysis plans and final statistical analysis plan, and will assist with data interpretation, and abstract and manuscript preparation. The PROSPECT organisational chart is in online supplementary appendix 2.
Supplemental material
Data monitoring committee
The PROSPECT Data Monitoring Committee (DMC) is independent from other persons involved in PROSPECT, and has the requisite expertise in randomised clinical trial design, epidemiology, biostatistics, warning guides/stopping rules, infectious diseases and critical care. The primary responsibilities of the DMC are to independently review reports prepared at the Methods Centre regarding: (1) recruitment (centre and patient) and screening, consent and coenrolment rates; (2) protocol procedures (randomisation, stratification, protocol adherence including maintaining blinding); and (3) data tables for two blinded interim and final analyses. After each interim analysis, the DMC will recommend whether to continue, suspend or terminate enrolment.
The roles and responsibilities of the DMC are as outlined and approved in the PROSPECT DMC Charter,22 modelled on the Data Monitoring Committees: Lessons, Ethics, Statistics (DAMOCLES) Study Group charter.66
Patient and public involvement
In PROSPECT, we are involving patients and their families in the following ways. First, before beginning PROSPECT, we ensured that patients and families were supportive of the use of probiotics in the ICU setting. We conducted a substudy nested within the PROSPECT Pilot16 17 whereby patient’s substitute decision makers were interviewed at the time of enrolment to explore their comfort with probiotic use during critical illness.67 In total, 103 SDMs participated in eight centres. We found no difference in characteristics of substitute decision makers who consented versus declined the PROSPECT Pilot. Rather, the rationale for substitute decision maker consent was related to personal beliefs regarding possible benefits to the patient, as well as predictions of patient’s wishes regarding this trial. These findings gave us confidence that patients and families were supportive of the trial. Second, in accordance with many local ICU research practices, patients who gain capacity after resolution of their critical illness are asked to agree to ongoing participation in the trial. We have no formal patient or family advisor for PROSPECT. When PROSPECT results are available following trial completion, we will ensure the results are disseminated by having the academic message of each ‘in press’ manuscript translated into press releases for the public. Possible target hospital media include newsletters, emails and intranet bulletins. High citizen awareness and probiotic consumption predict strong public interest.
Ethical oversight
PROSPECT is approved by Health Canada (#9427-M1133-45C), the REBs of all participating hospitals, and Public Health Ontario. The study is under way in accordance with Good Clinical Practices following the Tri-Council Guidelines68 and in accordance with ethical principles of the Declaration of Helsinki.69 Access to the database, study-related files and source documents for scientific or auditing purposes is possible during and after the trial for any PROSPECT Methods Center staff, Health Canada authorities or REB representatives (local, provincial or central). Participants will not be identified by name, and confidentiality will be maintained unless otherwise regulated. Data will be retained for 25 years as per Health Canada.
The DMC will use conservative ’warning guides' for apparent benefit in PROSPECT; there are no stopping guides for futility.
Funding
PROSPECT is funded by peer-reviewed grants (Canadian Institutes of Health Research, Canadian Frailty Network, Physician Services Incorporated, Hamilton Academic Health Sciences Organization, and Academic Medical Organization of Southwestern Ontario), and funds from St. Joseph’s Healthcare Hamilton and McMaster University. The study products are donated by the manufacturers of L. rhamnosus GG (i-Health) which have no role in the trial conception, design, conduct, oversight analysis or write-up.
Knowledge dissemination
Results of the trial and secondary manuscripts will be communicated through conventional academic channels (eg, abstracts, posters, peer-review manuscripts), at professional healthcare fora (eg, grand rounds, teaching sessions, in-services, quality improvement councils) and via media (eg, newspapers, radio, television, blogs, twitter, etc).
Discussion
Probiotics may be a simple, cost-effective strategy to prevent VAP.70 However, despite encouraging findings of efficacy, trials to date have been limited by insufficient power and risk of bias.9–11 Studies of probiotics in the critical care setting have been criticised as difficult to interpret due to differences in populations and heterogeneous probiotics and combination products used.71 Indeed, experts in the field have emphasised the need for well-powered studies of probiotics in the ICU setting.71 To address this call, PROSPECT is a large, international, rigorous multicentre randomised trial that aims to determine whether probiotics are effective, have no benefit or are harmful in critical illness.
Additional strengths of PROSPECT include representation of persons greater than 65 years of age to enhance the generalisability of the findings, and separate peer-review funding for this population from the Canadian Frailty Network.65 The efficacy of probiotics may be less in the elderly, as illustrated by a recent rigorous trial that found probiotics did not prevent C. difficile infection in Persons ≥65 years admitted to hospital and receiving at least one antibiotic.72 We are also documenting baseline pre-hospital frailty with the Clinical Frailty Score73 to further understand the relationship between frailty, immunosenescence and critical care-associated infections.
Given previous meta-analyses suggesting that probiotics may reduce all healthcare-associated infections in the ICU,9 we are evaluating all infectious outcomes in PROSPECT. Also, a recent large trial of 2556 healthy newborns conducted in rural India showed that synbiotics (Lactobacillus plantarum plus fructooligosaccharide) decrease the risk of sepsis and lower respiratory tract infections within 60 days.74 It is unknown whether the benefit was from the L. plantarum or the addition of fructooligosaccharide; however, these results suggest that modification of microbiota can reduce infections. Given the growing interest in the dysbiosis of critical illness, this trial will advance our understanding of whether microbiota modification with probiotics has any influence on infectious and non-infectious clinically important outcomes.75–77
The International Conference for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E978 and SPIRIT18 guidelines endorse a separate statistical analysis plan for clinical trials. Recommendations18 include more technical and detailed elaboration of the principal features of the analysis described in the protocol, including procedures for executing the statistical analysis of the primary and secondary variables and other data.78 We followed these expert recommendations19 for 55 items in six sections: Title and Trial Registration (11 items/subitems); Introduction (two items); Study Methods (nine items/subitems); Statistical Principles (eight items/subitems); Trial Population (eight items/subitems); and Analysis (17 items/subitems). Dissemination of this document aligns with calls to make statistical analysis plans publicly available79 to aid in the transparent reporting of trial results.
Trial status
PROSPECT is supported by a long-standing research consortium (the Canadian Critical Care Trials Group) dedicated to investigator-initiated, peer-review funded studies designed to understand and improve the outcomes of critically ill patients.21 Recruitment is ongoing, with 73% of the target sample size accrued as of May 2018. At the first interim analysis, the DMC made no suggestions to suspend enrolment. Randomisation is anticipated to continue until approximately April 2019. Final data entry, data validation and outcome adjudication will ensue for 6–9 months thereafter, with an anticipated database lock by December 2019, followed by the terminal statistical analyses. PROSPECT results will inform global practice in critical care medicine.
Acknowledgments
The trial was designed by the PROSPECT Steering Committee and improved by Dr Dawn Bowdish, the PROSPECT Investigators and Research Coordinators and the Canadian Critical Care Trials Group. We are grateful to other Methods Center staff for their expertise with PROSPECT data management, including Lois Saunders, Shelley Anderson-White, Alyson Takaoka, Mary Copland, Megan Davis and France Clarke. We wish to thank Drs Bram Rochwerg and Nick Daneman for their peer review of this manuscript.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
Footnotes
Contributors Concept and design: JJ, DH-A, LTh, DJC. Acquisition, analysis or interpretation of data: JJ, DH-A, LT, MM, JM, FL, EHD, NZ, DL, MS, DJC. Drafting of the manuscript: JJ, DH-A, LT, DJC. Critical revision of the manuscript for important intellectual content: everyone. Statistical analysis: DH-A (Trial Biostatistician), LT (Senior Biostatistician), DL, MS. Obtained funding: everyone. Administrative, technical or material support: DH-A, EHD, NZ, LT, DL, MS. JJ and DJC as co-principal investigators take responsibility for the integrity of the data.
Funding This work was funded by the Canadian Institutes of Health Research, Canadian Frailty Network, Physician Services Incorporated, Hamilton Academic Health Sciences Organization and Academic Medical Organization of Southwestern Ontario, as well as St. Joseph’s Healthcare Hamilton and McMaster University. iHealth is providing the blinded study product. FL is a recipient of a Research Career Award from the Fonds de la recherche du Québec-Santé. MS holds a Canada Research Chair in Interdisciplinary Microbiome Research. DJC holds a Canada Research Chair in Knowledge Translation in Critical Care.
Competing interests None declared.
Ethics approval Hamilton Integrated Research Ethics Board REB# 15-322.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Next of kin consent obtained.