Article Text

Abstract
Introduction Surveillance of unintended effects of pharmaceuticals (pharmacovigilance or drug safety) is crucial, as knowledge of rare or late side effects is limited at the time of the introduction of new medications into the market. Side effects of drugs may involve increased or decreased risk of cancer, but these typically appear after a long induction period. This fact, together with low incidences of many cancer types, limits the usefulness of traditional pharmacovigilance strategies, primarily based on spontaneous reporting of adverse events, to identify associations between drug use and cancer risk. Postmarketing observational pharmacoepidemiological studies are therefore crucial in the evaluation of drug-cancer associations.
Methods and analysis The main data sources in this project will be the Norwegian Prescription Database and the Cancer Registry of Norway. The underlying statistical model will be based on a multiple nested case–control design including all adult (~200 000) incident cancer cases within the age-range 18–85 years from 2007 through 2015 in Norway as cases. 10 cancer-free population controls will be individually matched to these cases with respect to birth year, sex and index date (date of cancer diagnosis). Drug exposure will be modelled as chronic user/non-user by counting prescriptions, and cumulative use by summarising all dispensions’ daily defined doses over time. Conditional logistic regression models adjusted for comorbidity (National Patient Register), socioeconomic parameters (Statistics Norway), concomitant drug use and, for female cancers, reproduction data (Medical Birth Registry), will be applied to identify drug-use–cancer-risk associations.
Ethics and dissemination The study is approved by the regional ethical committee and the Norwegian data protection authority. Results of the initial screening step and analysis pipeline will be described in a key paper. Subsequent papers will report the evaluation of identified signals in replication studies. Results will be published in peer-reviewed journals, at scientific conferences and through press releases.
- cancer risk
- drug safety
- pharmacoepidemiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Large-scale study of drug-use–cancer-risk associations by linking and using information from multiple registries in Norway.
The study design is a multiple nested case–control study comprising more than 2 000 000 individuals including all adult cancer cases diagnosed between 2007 and 2015 in Norway and 10 matched controls.
Adjustment for comorbidity, other drug use and socioeconomic factors, but: not one size fits all—lack of data on confounders for certain drug-cancer combinations.
Low power for some combinations of less frequent cancer types and drug groups.
Introduction
Rationale and evidence gaps
Surveillance of unintended effects of pharmaceuticals (pharmacovigilance or drug safety) is an important task, as knowledge of rare or late side effects is limited at the time of the introduction of new medications into the market.1 Side effects of drugs may involve increased or decreased risk of cancer,2 3 but these typically appear after a long induction period.3 4 The excess risk of breast cancer induced by use of combined oestrogen–progestin menopausal therapy becomes apparent only after 5–10 years of continued use,5 6 and chemoprevention of colorectal cancer with use of aspirin also requires many years of regular use.7 8 The suggested chemopreventive effects of statins, eg, against colorectal9 and prostate cancer,10 11 also derive from observations among long-term users of statins. The long latency of cancer development together with low incidences of many cancer types limit the usefulness of traditional pharmacovigilance strategies, primarily based on spontaneous reporting of adverse events, to identify associations between drug use and cancer risk. Furthermore, carcinogenic effects of drugs are not likely to be reported as adverse events, unless the drugs have a mechanism of action, which leads to suspicion. Postmarketing observational pharmacoepidemiological studies are therefore crucial in the evaluation of drug-cancer associations. Such studies have previously identified a range of important associations between drug use and cancer risk, eg, carcinogenic effects of immunosuppressants, menopausal hormone therapy and phenacetin.2 6
Studies based on large administrative databases have contributed to the discovery of drugs with carcinogenic effects.2 6 The availability of these sources for systematic assessment of associations between long-term use of prescription drugs and cancer risk provides unique possibilities for replication studies of the significant associations identified.
This study is based on high quality data from the Cancer Registry of Norway (CRN)12 and the Norwegian Prescription Database (NorPD) containing detailed individual-level information on all dispensions of prescription drugs purchased by individual persons at all pharmacies in Norway since 2004.13 The availability of these Norwegian high quality nationwide registries of drug dispensations and cancer incidence provides an outstanding opportunity to establish active surveillance of cancer risk associated with use of prescription drugs. A similar initiative has recently been established in Denmark14 and Sweden.15 Within the Kaiser Permanente Program in California drug-cancer screening has been performed during the recent decades.16 These drug-cancer screening studies differ both with respect to the number of drugs and cancer types included in the study and the models and methods applied. As there is not enough experience and no consensus on how to best perform this type of studies, there is an urgent need to further explore the feasibility of different (combinations of) models and methods for this type of pharmacoepidemiological studies. In addition to the accumulation of evidence of drug-cancer screening signals, we aim to contribute to the method development for drug-cancer screening studies.
Aims and hypotheses
The primary aim of this study is the identification of associations between prescription drugs and various types of cancer in a large-scale registry-based approach using existing Norwegian nationwide health and demographic registries. Thus, we propose a hypothesis-free screening approach to uncover drug-cancer associations that may, if validated and confirmed in other studies, indicate potential carcinogenic or chemopreventive effects of prescription drugs. We also aim to gain methodological insights into the use of healthcare databases as well as pharmacoepidemiological and statistical methods for the evaluation of associations between drug use and cancer risk on a large scale.
The public health perspective of identifying associations between drugs and increased cancer risk is obvious, and potential dismissal of suspected associations and reassurance of the safety of drug intake with respect to cancer risk is of great public health importance. The latter can reassure patients about the safety of essential drugs and promote their appropriate use. Furthermore, identification of potential chemopreventive effects of drugs also holds great value in the development of new targets for cancer prevention and treatment. This project will also establish knowledge with respect to study design, use of register data, multiple testing and statistical methodology within the field of pharmacoepidemiology, which is necessary to establish regular periodic drug-cancer screening in the future.
Methods and analysis
Study population and data sources
The entire Norwegian population is covered by the publicly funded healthcare system. Several national administrative and disease registries have been established that may be linked using a unique individual identification number assigned to all inhabitants of Norway. Thus, true population-based studies of disease may be efficiently conducted within the framework of the Norwegian registry system.
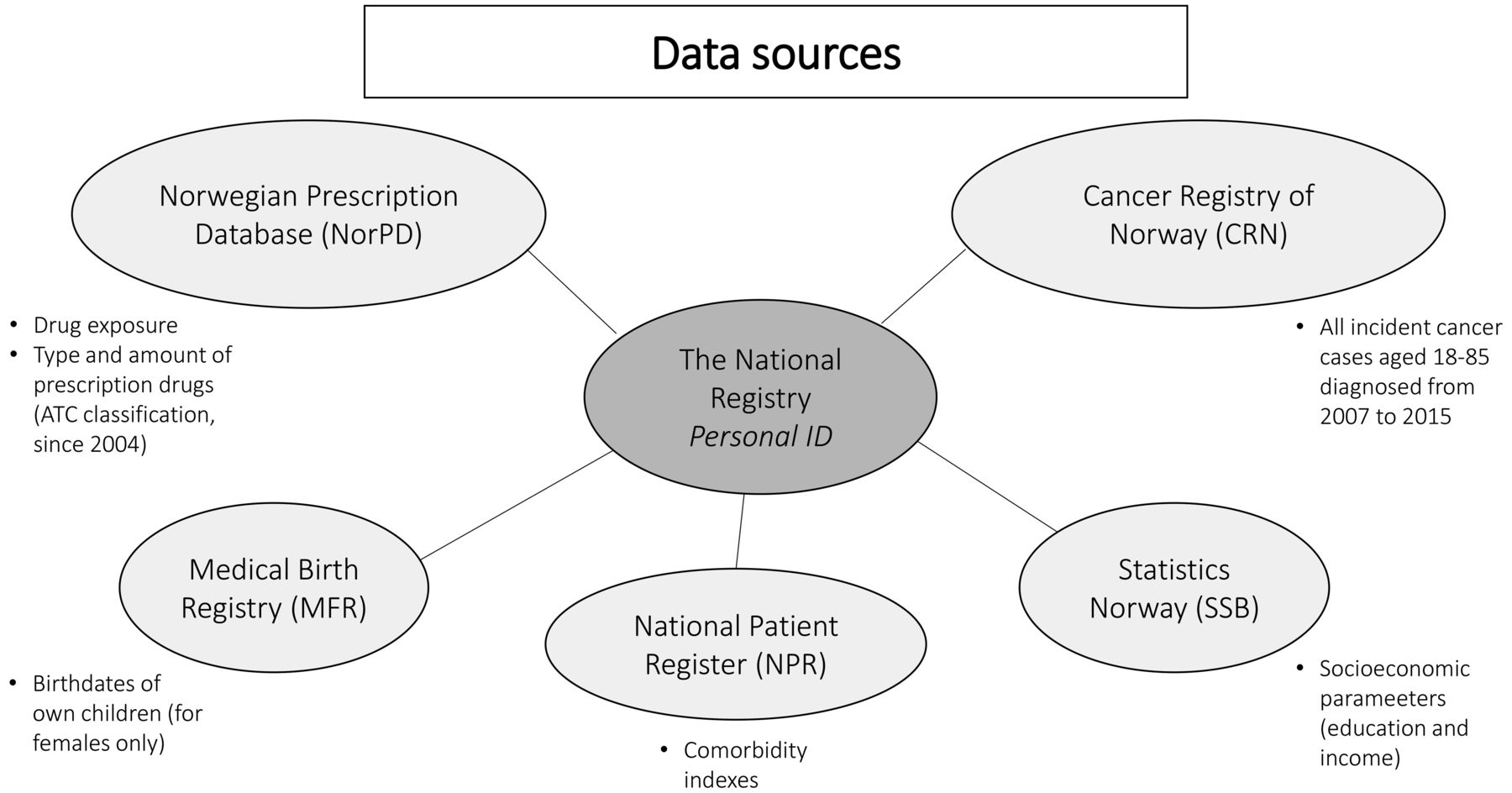
The main data sources in the proposed project will be the NorPD and the CRN (figure 1). The NorPD will provide drug exposure data. This database covers all prescription drugs dispensed to persons in ambulatory care in Norway since 2004 with information on the date of dispensing, amount dispensed and the type of drug which is classified according to the Anatomical Therapeutic Chemical (ATC) classification system.13 17 The CRN has recorded incident cases of cancer on a nationwide basis since 1953. The registry has been shown to have accurate and almost complete ascertainment of cancer cases.12 Furthermore, we will use data from the National Patient Registry (NPR), Statistics Norway (SSB) and the Medical Birth Registry of Norway (MBRN) to adjust for confounding factors, apply restrictions, and identify subgroups of special interest. Comorbidity indexes will be obtained from the NPR. Socioeconomic parameters (education and income) will be provided by SSB and reproduction data from MBRN.18 19

Data sources in the underlying study. ATC, Anatomical Therapeutic Chemical.
Study design and statistical methods
The underlying study will be implemented as a multiple nested case–control design20 based on all adult (aged 18–85 years) primary cancer cases (approximately 200 000) diagnosed from 2007 through 2015 in Norway as cases. Ten cancer-free controls will be individually matched (risk-set sampling) to each cancer case with respect to birth year, gender and index date (date of cancer diagnosis). Controls are thus cancer-free until index date, but might get cancer later in life. All cancer cases will be grouped topographically in ICD-10 categories (ICD-10: 10th revision of the international classification of diseases; as used in Cancer in Norway 21) and histopathological subgroups. Information from the CRN on the cancer diagnosis (date of diagnosis, morphology, etc) will be provided as well. The cancer case and its matched controls are referred to as a matched case–control set.
We chose a nested case–control design instead of the cohort design because the estimates from a conditional logistic regression of nested case–control data estimate the HR from the Cox regression using the entire cohort, furthermore with 10 controls per case the uncertainty of these estimates are only negligibly larger than the uncertainty of the estimates using the entire cohort. At the same time, a nested case–control design is more computationally efficient, and from an ethical perspective using a dataset not larger than necessary to answer the research question is preferred.
For each ICD-10 category and corresponding histopathological subgroups, associations between all second and fourth level drug classes of the ATC classification system and cancer risk will be evaluated. Each registered drug dispension contains information on the fifth level ATC drug class, date of dispension, type and amount defined daily dose (DDD) of the prescription. Thus, all feasible combinations of cancer and drug types are thus eligible for association testing (figure 2A). As a first step, we will focus on cancer types (and histopathological subtypes) with good power (>70% to detect HR=1.5) across different types of drugs.

{kind=link}
{kind=link}
Study design (A), included time period of drug dispensions (B) and exposure definition (C) in the underlying study. ATC, Anatomical Therapeutic Chemical; DDD, defined daily dose.
As a next step, we will retrospectively use information on the drug dispensions from index date back to 2004 for all individuals included in the study in order to quantify drug exposure. Figure 2B illustrates this for one fictive matched case–control set. For the drug type of interest, we start with information on dispensings of the drug type of interest from the index date back in time until 2004 when NorPD started individual registration. We thus exclude data from the last year before index date, because drug exposure the last year before cancer diagnosis is unlikely to influence the cancer development and because drug intake in this period might be due to treatment of first cancer symptoms.3 22 We also exclude all individuals with dispensings of the drug type of interest in 2004 in order to be able to determine the time point the individuals started with the drug intake. If a case needs to be removed based on this, the whole matched case–control set will be excluded from the analysis. The analysis will be done both including and excluding individuals with dispensings of the drug type of interest in 2004, thus investigating the impact of long-term users on the results.
Based on this, we will define drug exposure in two different ways: the crude exposure defining chronic users and non-users based on the number of prescriptions and the cumulative exposure based on the cumulative DDDs (figure 2C).
Crude exposure definition
Based on all dispensings within the time interval of interest (2005 to index date—1 year), drug use is grouped as non-users (0–1 drug dispensings), intermediate users (2–7 dispensings) and long-term users (at least 8 drug dispensings). We thus assume that eight prescriptions correspond to 2 years of drug use as drugs for chronic treatments are typically supplied for 3 months use at each dispensing.
Cumulative exposure definition
Based on all dispensings within the time interval of interest (2005 to index date—1 year), we will use the total number of defined daily doses (DDDs) across all dispensings as exposure to model a cumulative effect and establish possible dose-response relationships.
Statistical methods
We will investigate (screen) the whole dataset across all drug–cancer-type combinations for associations between drug use (exposure) and cancer (outcome). We will thus evaluate whether a drug is used disproportionally more often (or seldom) among cases compared with controls, which would indicate that the drug is associated with increased (or decreased) cancer risk. This approach does test for associations and does not imply approval or disapproval of causal effects. Moreover, this method is hypothesis-free, but potentially hypothesis generating.
The statistical analyses of all drug–cancer-type combinations will be performed based on matched-pair techniques, that is, by applying conditional logistic regression with drug use modelled as described above. This approach will automatically adjust for age, gender and calendar time, as these variables are matching variables. In addition, we will adjust for comorbidity measured by the Charlson Comorbidity Index23 and the Norwegian Patient Registry Index.24 We will also adjust for concomitant use of other prescription drugs, if the drug intake exceeds 2 years, and education, income and occupation registered in SSB. Moreover, we will adjust for the region of residence. This model is similar to the one used by Pottegård et al.14
Interesting signals for a specific drug–cancer-type combination will be defined based on the resulting p values (see also below), which combine information from the effect and sample size, variation in the data and the assumed (and adjusted) type-I-error. Interesting signals will be evaluated further in post hoc tests both with respect to relevant anatomical and histopathological subtypes of the relevant cancer-type category of interest, as well as the fourth and fifth level drug classes of the ATC classification system for the drug classes of interest.
Multiple testing
As described above, we will initially test for all possible associations between drug use on the second and fourth level of the ATC classification system and the topological and histological cancer type categories as described above. We assume the case–control studies to be independent across cancer types, while tests within each cancer type across drug types are based on the same study population and thus subject to multiple testing.
Obviously, in a hypothesis-generating approach like this, the chosen threshold should not be chosen too strict. On the other hand, resulting effect sizes or p values do not have an interpretation unless they will be compared with the underlying type-I-error by taking the amount and dependency structure of statistical tests performed into account. Otherwise, the screening of large numbers of associations would produce statistically significant associations purely by chance. We need to find a balance between a long list of interesting findings, which includes too many false positives and a short list of interesting signals where too many true positive signals have been removed. The way we solve this is by choosing a liberal overall type-I-error threshold and a multiple testing adjustment of the resulting p values, which allows the investigators to interpret the signal strengths. Moreover, all interesting findings will be subject to further analyses and internal validation (see below). The hypotheses in our approach are dependent as they have a hierarchical structure. For example, the second, fourth and fifth level of the ATC classification system have a hierarchical structure since a specific fourth level ATC code consists of a number of non-overlapping fifth level ATC codes. This hierarchical structure of hypotheses can be most effectively taken into consideration by Closed Test Procedures. These procedures are based on the closure principle25 and have become increasingly more popular as its application provides a foundation for most multiple testing methods used in clinical and pharmacoepidemiological applications.26 27
Follow-up analysis and future perspectives
Interesting signals from the screening of drug-cancer associations (as described above) do not only arise from true causal effects, and we will aim to filter out signals caused by biases like uncontrolled confounding or chance. In a subsequent internal validation step, we will filter the interesting signals to separate out those signals, which qualify for further investigation. In order to achieve this, we will combine biological, pharmacological and epidemiological knowledge with several approaches as explained by Pottegård et al.28 Thus, robustness against the choice of study design, the assessment of dose-response patterns and the distribution of the cancer risk over time as well as considerations of biological plausibility and possible uncontrolled confounding will be evaluated. For drug types usually given simultaneously with other drug types, similarities and interactions will be evaluated.
Signals which persist after the internal validation step are subject to external validation. We have an established collaboration with the Danish Cancer Society and the University of Southern Denmark, which has already a similar ongoing project based on the Danish Cancer Registry and the Danish Prescription Registry.14 Their results and the results generated from the drug-cancer screening within the Kaiser Permanente Program in California and the Swedish study15 16 will give us the possibility for replication. Thus, these data sources could be used to confirm any signals generated in the Norwegian setting together with a meta-analysis combining our results with those available from publications at that time-point.
Screening studies such as ours have the power to detect new drug–cancer-risk associations and will thus be hypothesis-generating, that is, examine possible effects of drug intake on subsequent cancer development. Causal relationships between drug use and cancer disease cannot be established by association studies alone, but interesting signals persisting after internal and external validation steps can be investigated in specifically designed follow-up studies where additional relevant confounders can be taken into consideration. Finally, when feasible, promising findings may be tested in animal or human in vitro studies, if suitable models are available.3
Power and sample size
The proposed screening study will include ~200 000 cancer cases and ~2 000 000 controls.
The power to calculate an association between drug use and cancer risk is dependent on the number of cancer cases (and controls) for a specific cancer type, the proportion of cases and controls using a specific drug and the effect size for this particular combination of drug use and cancer type. We will test for association of many different combinations of cancer types and drugs with each having different parameters for power calculations. We will give an impression of the expected power in this project by calculating the power for different combinations of the underlying parameters described above. For reasons of caution and simplicity, we assume a multiple testing adjusted type-I-error threshold of 0.2/10 000=2.10−5 in the underlying power simulation. The adjusted type-I-error threshold is reflecting the number of tests for all performed combinations of the second to fourth ATC levels and topological and histopathological cancer types. Since we will use a more sophisticated (and less stringent) multiple testing adjustment in the proposed study, our power estimates reflect a lower bound of the expected power. What we find is that the power is relatively low (<80%) for less frequent cancer types and small effect sizes (OR=1.2). Increasing the OR to 1.5 expands the number of cancer sites with sufficient power but the vast majority still has a power below 80%. The power is sufficient (≥80%) for the more frequent cancer types and both effect sizes investigated here (OR=1.2, OR=1.5). Breast cancer serves as an example for the more frequent cancer types and liver cancer as an example for the less frequent cancer types.
All adult individuals in Norway aged 18–85 years at cancer diagnosis will be included in the study. Thus, the power will be as high as feasible in a nested case–control design with 1:10 matching based on Norwegian data at the present date and will rapidly increase in the future as there will be more cancer cases and prescriptions available for the analysis.
Study strengths and limitations
As mentioned above, all individuals in Norway aged 18–85 years at cancer diagnosis will be included in the study together with 10 matched controls for each case, leading to a large study sample of more than 2 million individuals. The unique Norwegian personal identification number can be used to link data from several nationwide registries in order to assess information on drug use, cancer incidence, comorbidities and socioeconomic parameters for each individual included in this study.
Although taking age, sex, the date of cancer diagnosis (index date), comorbidities, concomitant drug use and socioeconomic parameters into consideration when evaluating possible drug-cancer associations, there are many possibilities for unknown confounders influencing these associations. Confounders can potentially generate spurious associations or mask real associations. The most obvious confounded associations (eg, drugs used for chronic obstructive pulmonary disease and lung cancer risk) will be excluded manually after the initial screening.
Reverse causation denotes the phenomenon that an association between drug intake and cancer risk does not imply that the drug intake causes cancer.29 It could also be that a drug is given to treat an early manifestation of an outcome, which is not yet diagnosed. There are numerous examples of reverse causation within cancer epidemiology, and not only for drugs which should be kept in mind in evaluation of confounding.22 We will address reverse causation by routinely disregarding all information on drug use during the last year before the cancer diagnosis. This is also well justified for biological reasons,3 since such recent exposure is unlikely to have contributed to the carcinogenesis.
For those drug-use–cancer combinations where surveillance of drugs users is more intense than for non-users, ascertainment bias plays a role, if the type of surveillance might lead to an (earlier) cancer diagnosis. This type of bias will partly be addressed by the adjustment for additional drug use and comorbidity. It will also be addressed in the internal validation step by excluding obvious pairs of drug-use–cancer-type combinations manually after the initially screening.
Patient and public involvement
As the study proposed by the present protocol is register-based, the research question and outcome measures were not influenced by any specific patient priorities, experiences or preferences. Rather, their formulation was based upon our own priorities for patient benefit and result interpretation. The nested case–control study described by the protocol uses only data from nationwide population-based registers and thus will not include a recruitment process for patients, who will not be involved in neither the design nor conduct of the study. All results will be distributed via the relevant patient and drug user groups, as well as peer-reviewed journals and scientific conferences. The study described by the present protocol is not a randomised control trial and will not have measures of intervention that could burden patients in any way assessable.
References
Footnotes
Contributors BKA prepared the study and the first version of the manuscript. NCS, JIM, GU, EW, HT, KBD, ØK, AP and SF contributed to the study design and reviewed and revised the protocol critically for important intellectual content, and approved the final versions. BKA is the guarantor.
Funding This project is funded by the Cancer Registry of Norway fund for research on cancer.
Competing interests None declared.
Ethics approval The study was approved by the regional ethical committee (2016/352 Identifikasjon av karsinogene og kjemopreventive effekter av reseptpliktige legemidler) and the Norwegian data protection authority. We also received permission from all involved registries, except for Statistics Norway which is pending.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.