Article Text

Abstract
Introduction Mycobacterial diseases are a significant source of disease burden, with Mycobacterium tuberculosis being the most common infectious cause of death worldwide. Given this, the emergence of antibiotic resistance in these species is of particular interest. By examining the epidemiology of mycobacteria in humans and cattle in an area of intense human–animal contact (the Ugandan cattle corridor [UCC]) and using a novel whole-genome sequencing technique to analyse organism diversity, this study will explore the role bidirectional transmission of mycobacteria plays in the local ecology, as well the significance of zoonotic Mycobacterium bovis in the human population.
Methods and analysis This ongoing study includes both a cross-sectional analysis of the UCC mycobacteria-positive population and novel laboratory-based research focused on differentiating the species causing M. tuberculosis complex-linked disease. We will use a third-generation sequencing platform (the MinION sequencer from Oxford Nanopore Technology), comparing data from a sample subset to the Illumina platform as a means of measuring viability of the MinION platform in this specific setting. Our full sample set will be sequenced on Illumina and the data will be used to perform epidemiological and phylogenetic analyses.
Ethics and dissemination Ethical approval was obtained from both the University of Minnesota IRB committee and the Ugandan National Council for Science and Technology—Research Ethics Committee. Samples were obtained after patients signed an informed consent indicating samples could be retained and used for research purposes. All samples are deidentified, with only basic demographic and geographic information being retained per national tuberculosis (TB) recording guidelines. Significant drug resistance results will be referred back to the local TB control officer to inform patient care. Final results of the study will be submitted to infectious disease-specific journals and will be submitted to the annual Infectious Diseases Society of America (IDSA) meeting.
- tuberculosis
- microbiology
- epidemiology
- molecular diagnostics
- public health
- tropical medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The study population includes both human and animal samples, increasing the odds of finding evidence of bidirectional human–animal transmission.
The human population selected is predetermined to be tuberculosis (TB)-positive (graded ‘high’ by GeneXpert).
Given the polymicrobial nature of the samples, the ‘time to data’ required for each sequencing run will likely be longer than for a pure TB culture.
The cattle corridor of Uganda, while useful for studying bidirectional human–animal transmission, may not be as generalisable to other populations as other regions of Uganda.
Quantifiying actual exposure to cattle will be difficult given the open grazing behaviours of cattle in the region, as well as the frequency and variety of animal contacts in daily life here.
Introduction
Mycobacterium tuberculosis complex (MTC) is a family of closely related pathogens. Two significant members of this complex are the human pathogen M. tuberculosis (MTB) and the largely animal pathogen Mycobacterium bovis. MTB is the causative organism in human tuberculosis (TB) infection, the most common infectious cause of death in adults worldwide, just above HIV.1 M. bovis, while seen most commonly in animals, is able to infect humans, often through consumption of unpasteurised milk. Though thought to be very uncommon, it has been shown to possibly be able to spread person-to-person through respiratory secretions, as well.2 In nations with a higher HIV burden, especially in those with limited access to HIV antiretroviral therapy, mycobacterial infection becomes an even larger problem due to increased rates of TB infection, conversion from latent TB infection to active disease, as well as increased severity of disease when immunocompromised.1 3 Given these factors, it is imperative that we continue to refine our understanding of MTC disease, the related epidemiology and treatment methods.
M. bovis is not distinguishable from MTB by routine tests that are readily available in low and middle-income countries (LMICs). Typically, species level identification requires culture (a slow, multiweek process) combined with immunohistochemical or nucleic acid level testing, both difficult with limited budgets and supplies.4 5 M. bovis is intrinsically resistant to pyrazinamide, a first-line anti-TB medication, though mutations in the gene encoding pyrazinamidase, the enzyme responsible for metabolising pyrazinamide into the active form, pyrazinoic acid.6 This drug resistance is not routinely tested for on the GeneXpert platform (a common method of detecting MTC diseases in both high-income nations and LMIC). Additionally, alternative drugs may not be readily available in many LMIC settings, making prevention of human M. bovis infection a more reasonable approach rather than adjusting therapies to more expensive and harder to get antibiotics.
Drug resistance, particularly multidrug resistance, is a looming problem with regard to mycobacterial infections. Multidrug-resistant TB (MDR TB) and extensively drug-resistant TB are quickly emerging diseases in LMICs, and have begun to appear in high-income nations, as well.1 7 While finding new therapies for these pathogens is critical, equally important is identifying and stopping the development of drug resistance in populations with high prevalence and disease burden. Drug exposure and inadequate therapy are certainly paths to drug resistance development, but there are other situations that are theorised to be key to this evolution, as well. We theorise that the back-and-forth transmission of MTB and M. bovis between cattle and people in areas of intense human–animal contact is one of the driving forces behind the emergence and maintenance of MDR TB in settings like the one in Uganda’s cattle corridor (UCC). Previous studies have postulated similar ideas, such as aminoglycoside usage in cattle leading to streptomycin resistance in M. bovis found in human disease.2 Ugandan cattle farmers have been reported to give their cattle antimycobacterial drugs due to a belief that the medications will cause their cattle to gain weight or become healthier, raising concerns that MTB or M. bovis passing between these cattle and people will have the opportunity to develop drug resistances. Nations that have taken strict measures to control M. bovis infection in their cattle and cattle products have seen a marked decline in the proportion of human mycobacterial disease due to M. bovis, down to the estimated developed world prevalence of 1%–3% of mycobacterial disease.8
Only a handful of populations have been the focus of epidemiological studies of M. bovis prevalence, with the most common risk factors identified as being of an advanced age, being an agricultural worker, consuming raw milk (or being old enough to have lived in the prepasteurisation era) or living in or immigrating from an area with higher M. bovis prevalence and/or no animal surveillance.9–13 Comparatively, there are limited data on such infections and risk factors in humans in Uganda, as most studies to date have focused largely on finding M. bovis disease in livestock.7 14–16 Most notably, Nalapa et al found an estimated 10% of cattle slaughtered in Mubende district, Uganda to have a TB-like lesions (though only 8.4% of these had identifiable MTC bacteria in the lesion).16 Given that M. bovis disease is commonly known to be spread by eating infected meat, drinking infected dairy or cohabitating with infected cattle, this represents a significant disease vector in Uganda. The intense level of human–cattle contact in the UCC (a region stretching across the country which contains around half of the nation’s cattle) makes communities here an ideal focus for studies of M. bovis.
To address both the need for a rapid, accurate diagnostic test that differentiates M. bovis and MTB, as well as to expand our knowledge of M. bovis epidemiology in what is suspected to be a high-risk population, we have decided to use the MinION sequencer from Oxford Nanopore Technology. The MinION system is a compact bioanalyser that uses a flow circuit and charge displacement to rapidly identify molecules and, in the case of DNA, to provide sequence data. These data can then be compared with a cloud database or to a local database for many different sorts of genetic analysis (speciation, marker identification and so on). In our study, we are trialling this technology as a way to perform on-site analysis and differentiate MTB from M. bovis in a fast, reliable manner. Not having to send the sample elsewhere for analysis will increase yield and will also help validate this system for future research use in the field. One of the most appealing features in an LMIC setting is the size: this system is highly portable, able to be carried in your pocket and only requires a universal serial bus (USB) connection to a laptop or other computer to run.17 This project will be used to validate this platform for use with MTC-positive samples in an LMIC setting and resulting data will be used for epidemiological studies.
Methods
This study began in July 2017 with initial sample collection and DNA extraction. It is ongoing with an end-date to be determined once we reach our target for analysed samples. Currently, samples are still being collected and DNA extracted while preparations are being made for sequencing at the study site in Mbarara, Uganda.
Study site
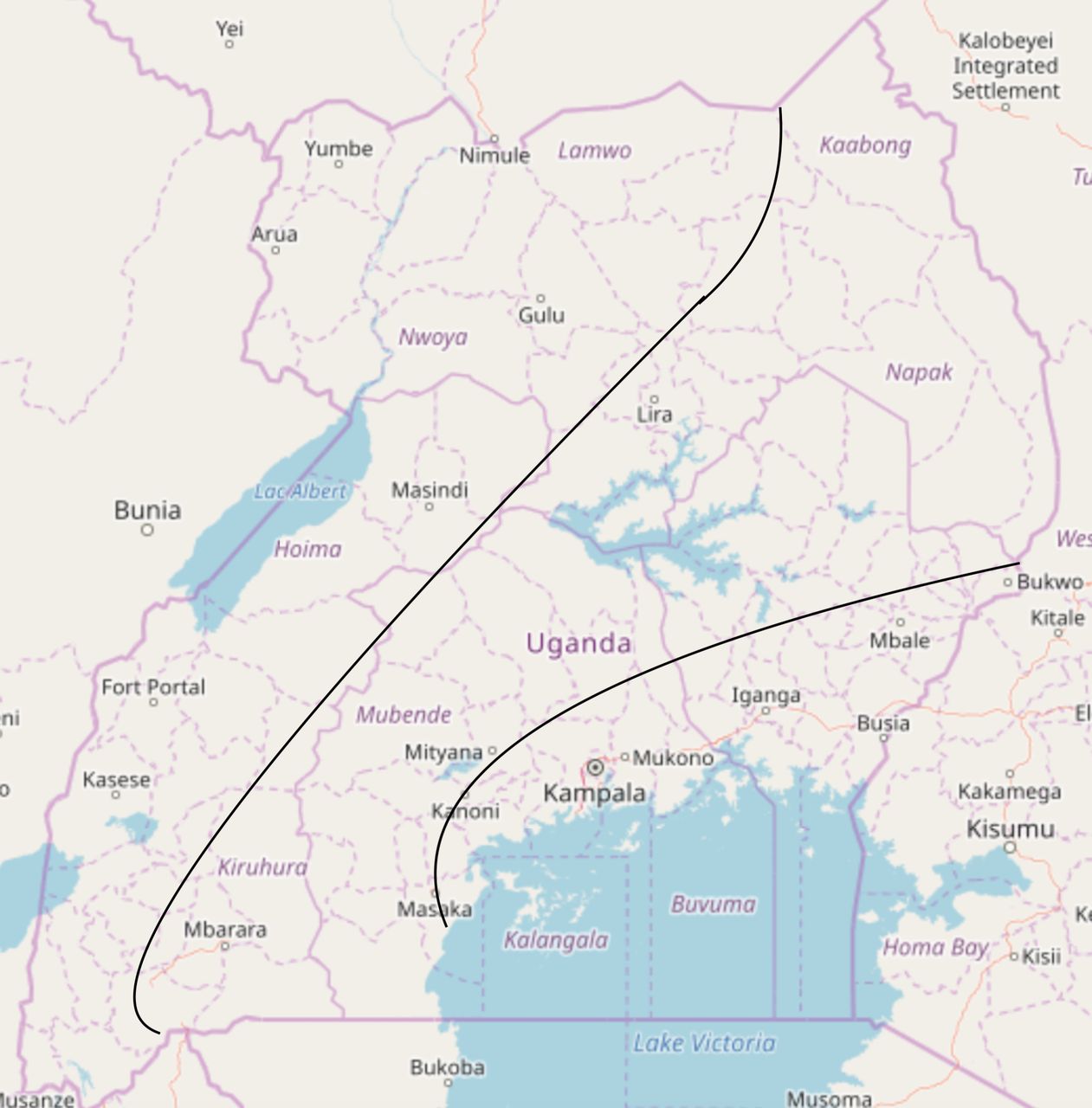
To focus our study on populations with intense human–cattle interaction, we have selected Mbarara, Uganda as our primary study site. Mbarara is the second largest city in Uganda and is located in the southwestern portion of the UCC (figure 1). The UCC is notable in that it contains a high density of cattle, representing about 45% of the nation’s cattle, increasing the level of contact between the people living here and the cattle and their food products.16 Additionally, Mbarara is home to Mbarara Regional Referral Hospital (MRRH) and Mbarara University of Science and Technology (MUST), both of which are collaborators on this project. MRRH serves as a healthcare hub for the region, treating both local patients and those sent from across the bulk of the southwest region from smaller medical centres. Due to this status as a referral centre, as well as the close relationship with MUST, there are more resources and equipment available at this site that we can use for our study.

Map of Uganda with the cattle corridor indicated as the region between the black borders. Also noted is the location of our study site, Mbarara, in the southwest. Original map obtained from Open Street Maps (© OpenStreetMap contributors, www.openstreetmap.org/copyright) and edited for demonstration and clarity purposes.
Study populations
We have identified and secured several samples sources for our study, which will allow us to look both at typical pulmonary MTC disease as identified by GeneXpert and extrapulmonary disease (detected by either GeneXpert or smear and culture) which is more commonly seen in M. bovis infection. Our primary sample source is MTC-positive sputum collected through the MUST clinical microbiology laboratory. This facility functions both as the clinical laboratory for MRRH and as a regional laboratory for surrounding district hospitals and community health centres in the southwestern Uganda region without the equipment or expertise needed to perform diagnostic microbiological tests. For the purposes of our study, we are retaining all specimens that produce a positive result graded as ‘high’ on the GeneXpert platform used by the lab.
Two other sources of human MTC-positive samples we are utilising are retained cerebrospinal fluid (CSF) from TB meningitis cases and acid-fast bacilli (AFB) positive blood cultures. The CSF samples, though likely lower in number than out other populations, will be useful as an extrapulmonary site for comparison to the sputum sample population. CSF from any TB meningitis case at MRRH will be collected and retained by MRRH and the MUST laboratory. The blood cultures being collected are part of separate study being conducted by MUST, MRRH and the MSF Epicentre examining AFB bacteremia in HIV-positive patients. Any samples not utilised by their study (due to exclusion criteria) will instead be used in ours, providing another extrapulmonary MTC disease population.
We have arranged for local slaughterhouses to retain suspect granulomas found in beef cattle, as well as blood and lymph node samples from the same animals. A significant number of animals are noted to have such granulomas, though only a portion will eventually grow mycobacteria. Once an animal is identified as potentially harbouring a mycobacterial infection, the above-mentioned samples will be collected and will then be transferred to our laboratory. They will then be tested for mycobacterial positivity via mycobacterial growth indicator tubes, with those that are positive being cultured and used for further genomic analysis as described below. Based on early sampling at these slaughterhouses, we are expecting two to three sets of samples (each set being from a unique animal) per week. The inclusion of these animal samples will allow us to look for possible links between animal and human mycobacterial disease in the two populations, both through similar drug resistance patterns and genotyping for isolate tracking.
In addition to providing both pulmonary and extrapulmonary diseases samples, sampling from several populations will also give us a collection of HIV-positive (blood cultures) and HIV-negative (majority of collected sputum and CSF) individuals. These samples are all catalogued by a unique identifying number along with basic demographic information (name, location of collection, residence of the patient), which will allow us to both acquire further information about samples of interest and perform analyses of risk factors for acquisition of M. bovis and/or antibiotic-resistant MTB strains. Other than the secured sample catalogue, all samples tubes and data collections are entirely deidentified.
DNA extraction and sequencing
All samples received by our lab will be processed in the same manner. After being analysed by GeneXpert, those samples that return positive and are graded high will be passed into our sample population. DNA extraction will be performed with two protocols initially: (1) Qiagen DNA Mini kit according to the manufacturer-provided manual and (2) the cetyltrimethylammonium bromide (CTAB)-based protocol described by the TB Alliance in the Shortening Treatment by Advancing Novel Drugs (STAND) trial laboratory practices manual, with minor modifications made due to our samples being clinical sputum samples rather than cultures.18 This CTAB protocol is described as follows. Briefly, sputum samples (containing the GeneXpert processing fluid) are centrifuged at 14 500 rpm for 15 min to pellet the cellular component. The supernatant is discarded, and the pellet is resuspended in 400 μL of TE buffer in a 1.5 mL microcentrifuge tube. The sample is then heat killed in a block set at 95°C for 30 min. After this, 50 μL of lysozyme (10 mg/mL in Tris-EDTA [TE] buffer) is added and the sample is incubated at 37°C overnight (typical range is from 18 to 24 hours). After this incubation, 70% of sodium dodecyl sulfate (10%) and 5 μL proteinase K (20 mg/mL) are added and the sample is incubated at 65°C for 15 min. A mixture of 10% CTAB and 0.7 M NaCl is prepared and heated to 65°C, with 100 μL of this heated mixture than being added to the sample tube along with 100 μL of 5 M NaCl. This sample mixture is gently pipetted and then incubated at 65°C for 10 min. Following this, 750 μL of chloroform:isoamyl alcohol (24:1) is added and the tube is mixed by inversion. After centrifugation at 14 500 rpm for 5 min, the aqueous phase is gently transferred to a new 1.5 mL microcentrifuge tube 450 μL of ice cold isopropanol and stored at −20°C for 30 min to an hour. This tube is then centrifuged at 14 500 rpm for 15 min (with attention paid to the orientation of the tube as the resulting pellet may be hard to see). The supernatant is removed, and the sample is washed with ice cold 70% ethanol. After a 5 min centrifugation at 14 500 rpm, the remaining ethanol is removed and the sample is allowed to air dry for 15 min. Finally, the pellet is resuspended in 50 μL of TE buffer and incubated at 65°C (to promote resuspension), and is then ready for downstream use.
These two extraction protocols will be compared for quantity and quality of DNA extracted via QuBit and sequence read length (as determined by the MinION sequencer), with the more favourable protocol being carried forward for the remainder of the study. This is key as there are very few studies on a standardised extraction protocol for clinical sputum samples containing MTB, and none looking at this situation when the sputum has already been processed for GeneXpert analysis.
Following extraction, the samples will be quantified using a QuBit fluorometric quantification device from ThermoFisher (catalogue number: Q33226). This quantification step is necessary due to input DNA requirements of the MinION platform. Once the DNA has been quantified and diluted to the proper concentration, DNA libraries are created for sequencing via the 1D Ligation Reaction Kit (SQK-LSK108) from Oxford Nanopore Technologies (with additional reagents purchased from New England Biosystems). This library preparation involves bead purification of DNA as well as dA-tailing ligation to allow the nucleic acid to bind to loading beads used by the MinION sequencer. After library preparation, the sample is loaded into an Oxford Nanopore Technology MinION flowcell (FLO-MIN106) and sequencing is performed via their proprietary software suite.
Further details of all kits and processes involved can be obtained by accessing the respective manufacturer’s website and using the item or catalogue number provided above.
Data analysis
Our target number for samples sequenced on the MinION platform is 50 samples (the exact proportion of sputum, blood, CSF and granuloma samples subject to sample availability), a number which should allow us to test the accuracy of the MinION platform across each type of sample with several unique samplings. For each sample sequenced on the MinION platform, we will sequence an aliquot from the same samples on the Illumina platform as means of comparison and validation of the MinION data, with each base pair of the sequenced serving as a chance to measure correlation between the data obtained from the two platforms. Further samples beyond the 50 MinION-sequenced portion will only be sequenced by Illumina due to lower cost per sample. Our goal for this exploratory study is to start with an initial target of 250 samples, divided among the animal and human populations, for our initial epidemiological analysis before pushing forward into a larger study. By sequencing at least 50 samples from aech population (animal and human) on both the MinION and Illumina platforms, we will be able to achieve a power of 0.95. If we estimate a low correlation between the platforms of 0.5, with a power of 0.95 and p<0.05, the required number of samples is 46, while higher correlation values bring the required number down significantly. Current correlation estimates from Ocford Nanopore are markedly >0.5, making our target of 50 samples sufficient to reach an appropriate power for the study.
Once all samples have been sequenced, we have several types of analysis planned for our data. First, we will use the data acquired from the MinION platform and perform ‘What’s In My Pot’ analysis using software provided by Oxford Nanopore Technology. This software analyses sequence data (both postsequencing and in real time) to determine the phylogeny of organisms found in the sample. This will be useful, given the polymicrobial nature of our clinical samples. It will also allow us to determine the coverage and length of sequencing run needed to obtain phylogenetic data on mycobacteria in these samples. Having a better understanding of necessary run time will help us adjust the protocol to make it more financially practical in an LMIC setting (shorter run times allow for more samples to be run per flowcell lifecycle). We will then compare the base-by-base calling of samples on the MinION platform to those called on identical samples on the Illumina platform as a measure of MinION accuracy. Beyond that 50 sample subset, we will use all our sequenced samples to examine the epidemiology of MTB and M. bovis in the UCC (figure 2 for full outline).

{kind=link}
{kind=link}
Flowchart summarising the data acquisition and analysis proposed in our study. MTBC, Mycobacteria tuberculosis complex; ABX, antibiotics.
Patient and public involvement
Samples being used for this study were collected as part of routine diagnostic care separate from our study and, as such, patients were not specifically involved in the study design since no additional burden, cost or risk was assessed to the patients. Results of the study will not be directly disseminated to patients, with the exception of clinically significant drug resistance data. These results will be relayed to the facility TB control officer. All data acquired in this study (epidemiological and sequence data) will be shared publicly in further publications.
Discussion
Our study aims to both validate a novel, rapid diagnostic tool for portable whole-genome sequencing in an LMIC setting and to then use this tool to explore the epidemiology of M. bovis diseases in a high-risk region. Initially, sequence data will be used to show fideility of the new MinION platform compated to the traditional Illumina platform when using clinical TB samples. We will also speciate the samples to examine the incidence of M. bovis in the UCC. Downstream from this, a future study will use these data to examine the role of animal–human bidirectional MTC transmission in the emergence of antibiotic resistance, as well as identify risk factors unique to the population for acquisition of M. bovis. Previous studies in other nations have laid the groundwork for our hypothesis that the level of animal contact in the UCC will increase the prevalence of human M. bovis disease, but until this sequencing technology, there has been no feasible way to confirm the hypothesis in the local setting.
Having a dataset of MTC from this region will serve as an excellent model to study the effects of bidirectional disease transfer between cattle and humans. Our inclusion of animal samples, as well as extrapulmonary and pulmonary human MTC-positive samples increases the generalisability of our data, as well as increase our chances of finding significant markers of such bidirectional transmission in both cattle and humans. Employing whole-genome sequencing will allow us to identify possible transmission linkages and check all known drug resistance loci. This information would be an important and impactful piece of evidence for agricultural officials, both in Uganda and in similar high-intensity contact regions, to focus on controlling animal TB as a means of reducing human M. bovis disease and slowing the emergence of drug resistance in MTC organisms.
In addition to the above-stated goals, this project will function as a transfer of technology and as a means of furthering our partnership with MRRH, MUST and the Ugandan government. These groups are vital if we are to stem a potential source of MTC drug resistance, a problem, that is, a worldwide threat, rather than an isolated one. The project goals also include teaching local researchers to use this new sequencing platform and bringing some supplies to allow for selective use of the platform. The process is straight forward and fairly quick, with many helpful resources available online from OxFord Nanopore. Currently, our estimated ‘per sample’ MinION sequencing cost is about US$200 when all reagents are included. This cost is potentially going to drop, however, as Oxford Nanopore continues to develop revised flow cells that may become partially reusable in the future. Though some of the reagents have temperature requirements while being transported, these are not any more stringent than typical enzymes or other eagents currently used in various labs throughout Uganda. By training and equipping labs in Uganda, we hope to have another ally in the fight against the emergence of drug-resistant TB.
We are confident that this project will yield important, convincing results. Using a novel, portable sequencing platform in our study, we feel that further validating the system will be a big step towards bringing full-genome sequencing to LMIC settings. Expanding our knowledge of MTC epidemiology in the UCC using a generalisable population will also aid further studies in similar populations, helping combat the emergence of drug resistance and stemming the transmission of animal TB to humans caring and working with them.
Ethics and dissemination
Samples obtained from other studies (blood) followed a similar standardised consent process. All samples are deidentified prior to being given to our laboratory, with only basic demographic (age, sex) and geographic (town or region) information being retained per national TB recording guidelines. Significant drug resistance results will be referred back to the local TB control officer to inform patient care. Final results of the study will be submitted to infectious disease-specific journals (Journal of Infectious Diseases, Tuberculosis, etc) and will additionally be submitted to the annual IDSA meeting (IDWeek).
Acknowledgments
We thank the NIH for their generous and continued support. We also thank Mbarara University of Science and Technology, Mbarara Regional Referral Hospital and the Mbarara MSF Epicentre for their partnership and collaboration. The corresponding author also thanks his mentorship team and coauthors, Drs David Boulware, Srinand Sreevatsan and Joel Bazira, for their expertise and advice throughout the project.
References
Footnotes
Contributors All authors contributed equally to the design of this protocol. MFP is the guarantor, being the primary data collector and sample processor. The corresponding author attests that all listed authors meet authorship requirements and that no others meeting the criteria have been omitted.
Funding Research reported in this publication was supported by the Fogarty International Center of the National Institutes of Health under Award Number 0437W009345. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclaimer The depiction of boundaries on the map(s) in this article donot imply the expression of any opinion whatsoever on the part of BMJ (or anymember of its group) concerning the legal status of any country, territory,jurisdiction or area or of its authorities. The map(s) are provided without anywarranty of any kind, either express or implied.
Competing interests All authors have completed the Unified Competing Interest form (available on request from the corresponding author) and declare: no support from any organisation for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
Ethics approval Ethical approval was obtained from both the University of Minnesota IRB committee and the Ugandan National Council for Science and Technology—Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.