Article Text

Abstract
Objectives Bimatoprost–timolol (bimatoprost 0.03%–timolol 0.5% fixed-dose combination [FDC]) and tafluprost–timolol (tafluprost 0.0015%–timolol 0.5% FDC) eye drops are currently the only topical intraocular pressure (IOP)-reducing therapies available as preservative-free (PF) prostaglandin and timolol FDC. The aim of this study was to investigate changes to ocular signs and symptoms when patients with ocular hypertension (OH) or open-angle glaucoma (OAG) switched from PF or benzalkonium chloride (BAK)-preserved bimatoprost–timolol to PF tafluprost–timolol eye drops.
Design This was a 12-week, open-label, phase IV study.
Setting Sixteen centres in Finland, Germany, Italy and the UK.
Participants Patients with OH or OAG (IOP on medication ≤21 mm Hg), treated with PF or BAK-preserved bimatoprost–timolol for ≥4 weeks before screening, and presenting with conjunctival hyperaemia and ≥1 ocular symptom.
Interventions Patients were switched to PF tafluprost–timolol once daily in the treated eye(s).
Primary and secondary outcome measures The primary endpoints were change from screening to week 12 in conjunctival hyperaemia and worst ocular symptom. The secondary outcome measures were changes from screening in ocular signs (other than conjunctival hyperaemia) and symptoms at week 12.
Results Of 123 enrolled patients, 121 were included in the intention-to-treat dataset, of which all were Caucasian and 54.5% were female; 76 patients used BAK-preserved bimatoprost–timolol and 45 used PF drops. Conjunctival hyperaemia and severity of worst ocular symptom following switch to PF tafluprost–timolol significantly reduced from screening to week 12 in all patients (p<0.001). The percentage of patients with ocular signs and symptoms was significantly reduced at week 12 compared with screening (p<0.001). IOP was not affected by the change of treatment.
Conclusions Switching from BAK-preserved or PF bimatoprost–timolol to tafluprost–timolol reduced both signs and symptoms of ocular surface disease with no clinically relevant effect on IOP.
Trial registration number EudraCT2014-005273-37; Results.
- glaucoma
- clinical pharmacology
- medical ophthalmology
- clinical trials
- intraocular pressure
- ocular surface
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The study allows for comparison between the effects of preservative-free (PF) tafluprost–timolol and both benzalkonium chloride (BAK)-preserved and PF bimatoprost–timolol formulations.
The study was conducted across 16 centres, limiting bias.
As this was not a randomised-controlled trial, there was a potential for selection bias; however, a randomised design would have been unethical as patients would have been required to adhere to medication that caused them notable ocular intolerance.
An open-label design could not be avoided for this study because the packages of BAK-preserved and PF bimatoprost–timolol and PF tafluprost–timolol were not identical.
Regression to the mean should be considered when interpreting the results.
Introduction
Glaucoma is a disorder often associated with elevated intraocular pressure (IOP) which, if left untreated, leads to retinal ganglion cell death, thinning of the retinal nerve fibre layer, optic nerve damage and cupping of the optic disc.1 In 2013, approximately 64.3 million people were affected by glaucoma, and the number is expected to grow to 111.8 million by 2040.2
Medical treatment of ocular hypertension (OH) and open-angle glaucoma (OAG) focuses on the long-term control of IOP.3 4 Several categories of IOP-lowering topical drugs are available including prostaglandin analogues (PGA) (generally the first-line treatment), β-adrenergic blockers, carbonic anhydrase inhibitors, α-adrenergic agonists and miotics.1 The greatest reduction of IOP is obtained with PGAs (25%–35%) followed by non-selective β-blockers (20%–25%), such as timolol; however, when patients fail to achieve IOP targets with monotherapy, fixed-dose combinations (FDCs) should be considered.5
There are only two available PGA–timolol preservative-free (PF) FDCs; PF bimatoprost 0.03%–timolol 0.5% (bimatoprost–timolol) and PF tafluprost 0.0015%–timolol 0.5% (tafluprost–timolol).6 Despite numerous comparative efficacy studies to date, few have compared different PF PGA therapies.7 While non-selective β-blockers, such as timolol, can cause bradycardia, arrhythmias and reductions in blood pressure, PGAs lack systemic side effects1 but may be associated with distinctive ocular adverse events (AEs), such as conjunctival hyperaemia.6 Preservatives such as benzalkonium chloride (BAK) are toxic to the ocular surface and may aggravate the signs and symptoms of ocular surface disease (OSD).8 In a recent study, conjunctival hyperaemia occurred at similar rates in BAK-preserved and PF bimatoprost–timolol-treated patients, suggesting that bimatoprost may have caused these AEs rather than the preservative.9 Additionally, switching from BAK-preserved bimatoprost monotherapy to PF tafluprost monotherapy has been shown to significantly reduce the severity of conjunctival hyperaemia.10 The objective of the present study was to evaluate the changes in ocular signs and symptoms in patients diagnosed with OH or OAG who were treated with BAK-preserved (0.005%) or PF bimatoprost–timolol eye drops (bimatoprost 0.03%–timolol 0.5%; Allergan, Irvine, CA, USA) prior to the start of the study and switched to PF tafluprost–timolol eye drops (tafluprost 0.0015%–timolol 0.5%; Santen Pharmaceutical Co., Ltd, Osaka, Japan).
Materials and methods
Study design
This was an open-label, phase IV clinical study (EudraCT registration number: 2014-005273-37) conducted at 16 centres in Finland, Germany, Italy and the UK from June 2015 to May 2016. The study was reviewed and approved by the appropriate Independent Ethics Committees in the participating countries and conducted in accordance with the Good Clinical Practice guidelines of the International Council on Harmonisation and the ethical principles of the Declaration of Helsinki.
Patient population
Patients included in this study were aged ≥18 years, diagnosed with OH or OAG, inclusive of both primary OAG and pseudoexfoliation glaucoma, and treated with bimatoprost–timolol in the evening (BAK-preserved or PF single-dose formulation) in one or both eyes for ≥4 weeks before screening. Patients presented at screening with conjunctival redness/hyperaemia of at least moderate severity (grade ≥2) in at least one treated eye and ≥one ocular symptom of at least mild severity (grade ≥2) in either eye. Exclusion criteria included: use of more than two active medicinal agents to treat OH or OAG in the 6 months prior to screening; anterior chamber angle grade <2 (Shaffer classification) in either treated eye; and any corneal abnormality or other condition preventing applanation tonometry, including prior refractive eye surgery and IOP >21 mm Hg in the treated eye(s) at screening. A full list of inclusion and exclusion criteria is presented in online supplementary table S1.
Supplementary file 1
Treatments and assessments
Eligible patients had used BAK-preserved or PF bimatoprost–timolol (bimatoprost 0.03%–timolol 0.5%) eye drops in the evening for ≥4 weeks prior to screening. Study treatment kits, containing PF tafluprost–timolol eye drops (tafluprost 0.0015%–timolol 0.5%) in unit-dose containers, were dispensed to patients at the screening visit. Patients were not blinded to treatment because an open-label design could not be avoided owing to differences in packaging between BAK-preserved and PF bimatoprost–timolol and PF tafluprost–timolol. Each patient instilled one drop of tafluprost–timolol once daily at 21:00 (±1 hour) in the affected eye(s) for 12 weeks. Drug accountability documentation and dosing data from case report forms were used to assess treatment compliance. Patients were assessed at screening, and at 2, 6 and 12 weeks postscreening. After week 12, a poststudy visit was scheduled, and the investigator was free to prescribe any IOP-lowering medication.
Ocular signs and symptoms
Ocular signs and symptoms were assessed at every visit and were defined by the criteria described in online supplementary table S2. Ocular symptoms were evaluated per patient and treated eyes were considered together. Ocular signs were analysed in the worst eye designated for each sign at screening. The coprimary endpoints were changes from screening in conjunctival hyperaemia and worst ocular symptom at week 12. The severity of conjunctival hyperaemia was assessed from screening through to week 12. Use of the Ora CalibraTM Redness Scale #6.0 (0–4 scale) was made under licence from Ora, Inc. Patients indicated their perceived worst ocular symptom at screening.
Secondary endpoints were changes from screening in ocular signs and symptoms, other than conjunctival hyperaemia, at week 12. The patient was asked about each symptom by a leading question, with symptoms graded 0 (none), 1 (trace), 2 (mild), 3 (moderate) or 4 (severe). A total symptom score (0–20) was calculated. Fluorescein tear break-up time was assessed by examination of tear film under a slit lamp following instillation of 2 µL of non-preserved 2% sodium fluorescein dye to the eyes. The time taken (in seconds) to form micelles or for dry spots to develop was recorded as the break-up time. Corneal and conjunctival fluorescein staining were also evaluated. Using reference pictures (Oxford Grading scale) the corneal fluorescein staining and nasal and temporal conjunctival fluorescein stainings were scored from 0 to V each. The presence of blepharitis was also evaluated, and the severity was graded 0 (none), 1 (mild), 2 (moderate) or 3 (severe). Tear production was assessed using the Schirmer-I test for 5 min without anaesthesia.
Adverse events
Treatment-emergent ocular and non-ocular AEs were reported at each postscreening visit. The information obtained included event term, report source, the seriousness of the event, onset and resolution date, frequency, severity, relation to study drop instillation, location (left/right eye, both or not applicable), study drug treatment action, and the investigator’s causality assessment of the study treatment and outcome. All AEs were coded using the latest Medical Dictionary for Regulatory Activities.
Ocular safety and quality of life
At each visit, IOP was measured in both eyes using Goldmann applanation tonometry; the right eye was measured first. Two consecutive measurements were taken to determine the mean IOP. If the initial two measurements differed by ≥3 mm Hg, then a third measurement was taken and the median IOP was determined. Other measures of ocular safety and quality of life (QOL) are described in the online supplementary information.
Sample size
A mean change of 0.37 units (SD 1.12) from screening in conjunctival redness/hyperaemia was assumed from the previous tafluprost switch studies.11 Using these estimates, it was determined using a paired t-test that 100 patients would be required for a power of 90% for conjunctival hyperaemia and >99% for worst ocular symptom.11 12 The calculations were done using the nQuery Advisor (V.6.0). The paired t-test was initially used to calculate sample size. However, the Wilcoxon signed rank test was found to perform better for heavy-tailed distributions and was thus used for the primary analysis.
Statistical methods
The intention-to-treat (ITT) dataset included all enrolled patients who received at least one dose of tafluprost–timolol and had at least one postscreening primary outcome measurement available. The safety set included all enrolled patients who had at least one dose of study treatment and had a subsequent safety measurement. The primary outcome measures for ocular signs and symptoms were assessed using the Wilcoxon signed rank test. No imputations for missing data were carried out. However, sensitivity analyses using the last observation carried forward imputation were carried out for the primary outcome measures. The analyses of secondary outcome and IOP measures were completed using standard statistical methods for paired data (eg, McNemar’s test for binary data, Wilcoxon signed rank test for ordinal data and the paired t-test for continuous data). For AEs, both patient and event counts were calculated, and events leading to discontinuations were summarised. Best corrected visual acuity, biomicroscopy, ophthalmoscopy, visual field test, drop discomfort and Comparison of Ophthalmic Medications for Tolerability (COMTol) are ocular safety and QOL outcomes, which were analysed descriptively.
Patient involvement
No patients were involved in setting the research question or the outcome measures, nor were they involved in the design or implementation of this study. There are no plans to involve patients in the dissemination of results as the open-label nature of the study meant that patients were aware of which medication they received.
Results
Patient demographics and baseline characteristics
A total of 126 patients were screened. Of the 123 patients enrolled, two had no postscreening data and were excluded; therefore, 121 (98.4%) patients were included in the ITT analysis, of which 114 (94.2%) patients completed the study (BAK-preserved, n=71; PF, n=43) (figure 1). The safety set comprised 123 patients. The mean (range) age was 66 (36–86) years, and more than half of the patients were female (54.5%) (table 1). Of the patients, ~70% in both BAK-preserved and PF subgroups had used bimatoprost–timolol for at least 6 months; 76 patients had used BAK-preserved (62.8%) and 45 had used PF (37.2%) bimatoprost–timolol. Approximately 20% of patients were diagnosed with OH and 75% with OAG. Most patients (91.7%) required treatment in both eyes.
Demographics and baseline characteristics of enrolled patients

Patient disposition by previous bimatoprost–timolol treatment. After initial screening, three patients did not meet the inclusion criteria. A total of nine (7.4%) patients discontinued the study; five discontinued because of AEs and four withdrew from the study. AE, adverse event; BAK, benzalkonium chloride; PF, preservative-free.
Changes to signs and symptoms
A significant improvement was observed in the severity of both conjunctival hyperaemia and worst ocular symptom compared with screening after switching from bimatoprost–timolol to tafluprost–timolol (p<0.001 at weeks 2, 6 and 12). The mean ±SD deviation grade of conjunctival hyperaemia for all patients decreased from 2.26±0.45 at screening to 0.94±0.64 at week 12 (a mean reduction of 58.5%) (figure 2A). The percentage of patients with conjunctival hyperaemia significantly reduced from 76 (100%) and 45 (100%) patients at screening in BAK-preserved and PF bimatoprost–timolol groups, respectively, to 47 (66.2%) and 31 (72.1%) at week 12 (figure 2B). All patients identified a worst ocular symptom at screening, which was at least mild in severity; the number of patients with the identified symptom was reduced to 47 (41.2%) at week 12. The number of patients with moderate and severe worst ocular symptom decreased from 62 (51.2%) and 12 (9.9%) at screening, to 11 (9.6%) and one (0.9%) at week 12, respectively (figure 2C). In the BAK-preserved and PF bimatoprost–timolol subgroups, the number of patients with moderate and severe worst ocular symptom decreased from 46 (60.5%) to five (7.0%) patients and from 28 (62.2%) to seven (16.3%) patients, respectively (figure 2D).

Comparison of week 12 outcomes with screening in conjunctival hyperaemia and worst ocular symptom after switching from bimatoprost–timolol to tafluprost–timolol (A) change in conjunctival hyperaemia from screening (n=121) to week 12 (n=114); (B) breakdown of changes in conjunctival hyperaemia severity by subgroup at week 12 compared with screening. One patient in the ITT dataset violated inclusion criterion 2 and only had mild conjunctival hyperaemia at screening; (C) severity of worst ocular symptom at screening and week 12 in all patients; and (D) changes in severity of worst ocular symptom by subgroup at week 12 compared with screening. BAK, benzalkonium chloride; ITT, intention-to-treat; PF, preservative-free.
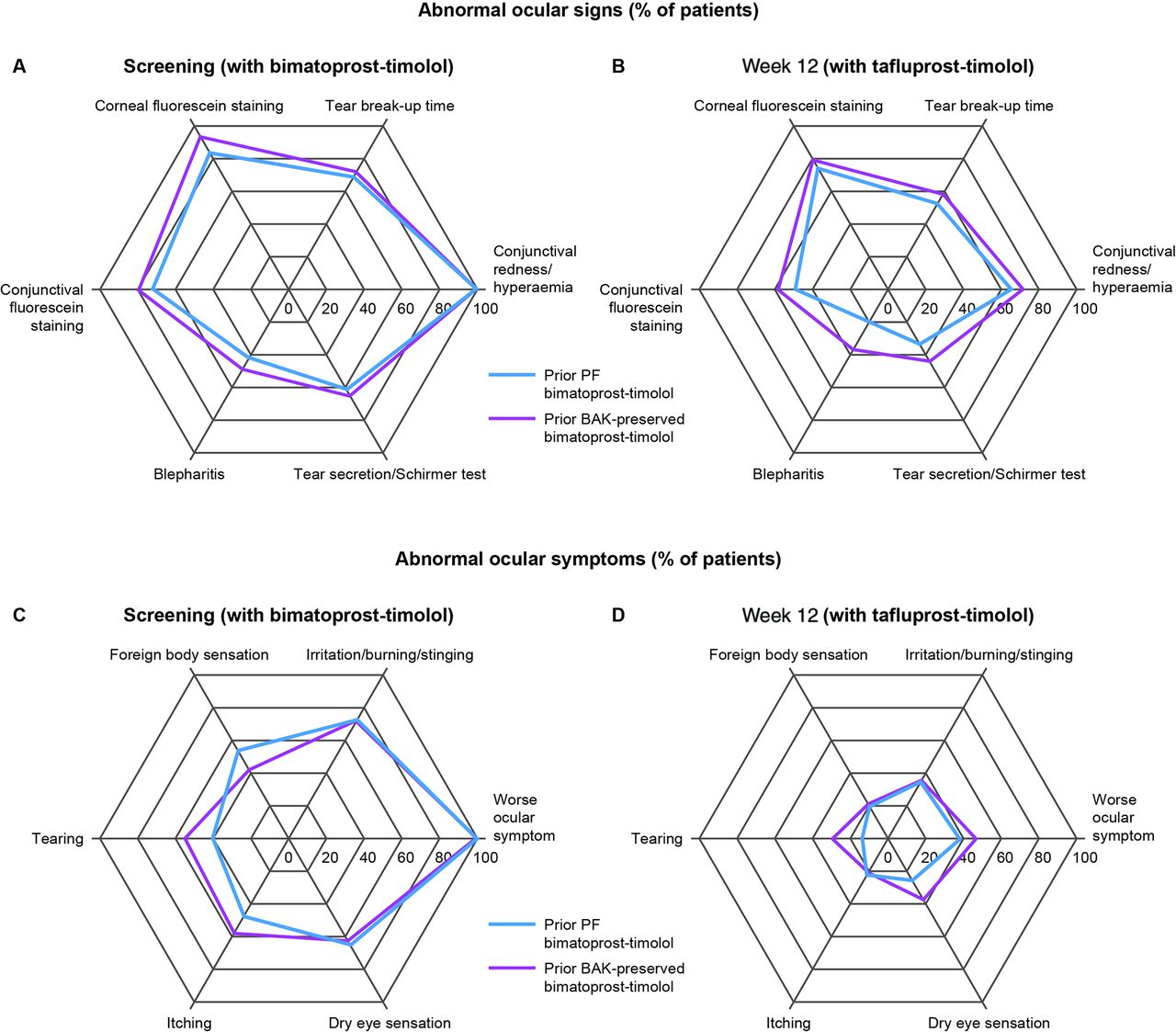
The frequencies of abnormal ocular signs and symptoms were significantly reduced at week 12 after switching from bimatoprost–timolol to tafluprost–timolol (p<0.012 for signs and p<0.001 for symptoms) (figure 3A, B, C and D). For ocular signs, the greatest relative reductions from screening were observed in tear secretion (screening, 62.8%; week 12, 37.7%) and blepharitis (screening, 44.6%; week 12, 27.2%). For ocular symptoms, all relative reductions were over 50%, and the greatest were observed in foreign body sensation (screening, 49.6%; week 12, 20.2%) and itching (screening 51.2%; week 12, 21.9%).
{kind=link}
{kind=link}
{kind=link}
Secondary endpoints (A) abnormal ocular signs at screening (n=121); (B) abnormal ocular signs at week 12 (n=114); (C) abnormal ocular symptoms at screening; (D) abnormal ocular symptoms at week 12. BAK, benzalkonium chloride; PF, preservative-free.
Adverse events
Overall, 70 treatment-emergent AEs based on the safety dataset (n=123) were reported by 41 (33.3%) patients during the study, of which 15 events in 12 (9.8%) patients were ocular and 55 events in 34 (27.6%) patients were non-ocular (table 2). Only 12 AEs in 10 (8.1%) patients were classified as being related to tafluprost–timolol. Two patients had serious AEs: worsening of arterial branch occlusion (resolved after 4 weeks) and paroxysmal atrial flutter with high-grade atrioventricular block (resolved in 2 days); both of which were adjudicated by the investigator and sponsor to be unrelated to tafluprost–timolol treatment. A total of five patients discontinued the study because of AEs, which were: two cases of moderate increase in IOP; moderate pruritus and eye pruritus, a moderate urticaria; and a severe increase in lacrimation. There were no deaths during the study.
The occurrence of related and unrelated ocular and non-ocular AEs in patients (n=123) after switching from bimatoprost–timolol to tafluprost–timolol
Ocular safety and quality of life
At screening, IOP was well controlled with bimatoprost–timolol treatment (n=123; mean IOP 15.9±2.1 mm Hg); this was sustained at week 12 (n=114; mean IOP 16.3±2.3 mm Hg) and was clinically insignificant and statistically non-inferior compared with screening (0.34 mm Hg; 95% upper limit 0.86 mm Hg; p=0.134). IOP was maintained at ≤21 mm Hg for >97% of patients and ≤18 mm Hg for >80% of patients. Other results for ocular safety and QOL are described in the online supplementary information (online supplementary table S3).
Discussion
Medical treatment of glaucoma aims to maintain patients’ visual function and QOL; however, nearly all patients with glaucoma will require a combined therapy to attain a greater than 30% 24-hour IOP reduction.13 14 Currently there are only two PF prostaglandin–timolol formulations available: PF bimatoprost–timolol and tafluprost–timolol. In this study, the effects of switching from bimatoprost–timolol to tafluprost–timolol on signs and symptoms of OSD and the effect of these changes in QOL were evaluated.
The study met both coprimary endpoints showing significant improvements in conjunctival hyperaemia and worst ocular symptom from screening to week 12. No statistical evidence of heterogeneity in the occurrence of ocular signs and symptoms was found between prior BAK-preserved and PF bimatoprost–timolol. This study has shown that patients receiving bimatoprost–timolol who present with signs and symptoms of OSD benefit from switching to tafluprost–timolol. Control of IOP was maintained, and there were no reports of unexpected AEs related to tafluprost–timolol or significant findings in ocular safety during the study. In this study, both the preserved and PF groups experienced a reduction in overall signs and symptoms. These results agree with a previous study, which found that the severity of both conjunctival hyperaemia and punctate keratitis was significantly higher with bimatoprost than tafluprost (p<0.02 and p<0.04, respectively).10 Additionally, these results agree with a previous double-blind phase III study where no significant differences in safety and tolerability between BAK-preserved and PF bimatoprost–timolol were observed except for more frequent skin pigmentation with PF bimatoprost–timolol.9 The observed ocular surface abnormalities and improved tolerability may thus be related to the prostamide-mimetic properties of bimatoprost as opposed to the preservative component of the bimatoprost–timolol formulation. However, there are studies that suggest that BAK may have a significant role in causing tolerability issues with PGA monotherapy despite once-daily dosing. A recent meta-analysis of two phase III studies including 339 patients who had switched from BAK-preserved latanoprost to PF tafluprost resulted in significant reductions in ocular signs and symptoms.11 Timolol treatment has been shown to induce only minimal hyperaemia or irritation in the eye.6 In this study, the percentage of symptom-free patients increased by week 12 concomitantly with improved ocular tolerability as reported in the COMTol questionnaire after switching from bimatoprost–timolol to tafluprost–timolol. This agrees with a previous study that found an association between advanced OSD and poorer glaucoma-related QOL than in patients without OSD.15 The aforementioned study also reported that OSD was associated with higher daily doses of BAK.
This study had several limitations. This was not a randomised controlled trial, and there was a potential for selection bias; however, a parallel-group (randomised) design with bimatoprost–timolol was considered unethical, because the patients would have continued using medication that caused them notable ocular intolerance. An open-label design could not be avoided for this study because the packages of BAK-preserved and PF bimatoprost–timolol and PF tafluprost–timolol were not identical. IOP readings were unmasked and may also have been subject to bias. ‘Regression toward the mean’ is the observation that if a variable is extreme on the first measurement, it will tend to be closer to the average on its second measurement. This may have introduced reduction in some ocular signs and should also be considered in the interpretation of these results. Compliance is likely to be higher in a study setting, and so these results may not be reflective of a real-world setting. Treatment persistence could not be investigated thoroughly because PF tafluprost–timolol was only commercially available for 26 patients in two of the participating countries (UK and Finland) at the time of the study.
In conclusion, switching from bimatoprost–timolol to tafluprost–timolol yielded clinical benefits in the presence of signs and symptoms of OSD in patients with OH and OAG over 12 weeks. Tafluprost–timolol provides a potential alternative treatment option for patients with OH or OAG.
Acknowledgments
Medical writing support was provided by Sandra Callagy, MSc, Helios Medical Communications, Alderley Park, Cheshire, UK, which was funded by Santen. The authors would like to acknowledge research nurses Helena Ollikainen and Helvi Käsnänen, Department of Ophthalmology, Kuopio University Hospital, Kuopio, Finland.
References
Footnotes
Contributors RRAB, KK, KL and CET contributed to the conduction of the study. JV contributed to the study design, statistical analyses and regulatory writing of the study. AR contributed to the study design, direction and monitoring. All authors contributed towards the writing of this publication.
Funding This work was supported by Santen.
Competing interests RRAB has received travel expenses from Santen. KK has received a consultant fee from Santen for an advisory board. KL has no conflicts of interest to report. CET has received department funding for the conduction of this study, personal fees and non-financial support from Santen not related to this study or manuscript, and department funding from Novartis and Allergan. JV has received fees for statistical services from Santen. AR is an employee at Santen Oy.
Ethics approval The study protocol was reviewed and approved by Independent Ethics Committees and national competent authorities in each participating country. The study adhered to the International Council on Harmonisation Good Clinical Practice guidelines and provisions of the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available. Please contact corresponding author at rb@rupertbourne.co.uk.
Patient consent for publication Not required.