Article Text

Abstract
Introduction Peritonitis is a major complication of continuous ambulatory peritoneal dialysis (CAPD), the risk of which is significantly influenced by the type of PD transfer system. Although the Y-disconnect and double-bag system is more efficient in preventing peritonitis compared with the spike system, little information is available to differentiate risks between different brands of the Y-disconnect double-bag system. A randomised controlled trial to evaluate the safety and efficacy of a newly introduced system is needed to provide the necessary clinical evidence to guide policy decision-making.
Methods and analysis The study is an open-label randomised controlled trial. A total of 434 patients with end-stage renal disease undergoing CAPD will be enrolled and randomised to either the intervention group, Stay Safe Link, or the control group, Stay Safe. All study subjects will be followed up and monitored for 1 year. The primary safety outcome is the rate of peritonitis while the primary efficacy outcomes are the delivered dialysis dose and ultrafiltration volume.
Ethics and dissemination The study was approved by the Medical Research Ethics Committee, National Institute of Health Malaysia. A written informed consent will be obtained from all participating subjects prior to any trial-related procedure and the study conduct will adhere strictly to Good Clinical Practice. The findings will be disseminated in a peer-reviewed journal.
Trial registration number NCT03177031; Pre-results.
- dialysis
- end stage renal failure
- peritonitis
- medical device
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The findings of the trial are likely to add to the existing literature the importance to assess peritonitis risk when interchanging between two Y-disconnect continuous ambulatory peritoneal dialysis (CAPD) systems.
The trial result will be useful in facilitating risk–benefit assessment and inform the registration of new Y-disconnect CAPD systems in Malaysia.
The study investigators, clinicians and study subjects cannot be blinded to treatment allocation.
The sample size calculation was performed based on a minimum 1.5 times increase in peritonitis rate ratio observed over 12 months and may not be sufficiently powered to detect any smaller differences.
Introduction
Background and rationale
Continuous ambulatory peritoneal dialysis (CAPD) is the predominant form of home dialysis treatment for patients with end-stage renal disease (ESRD). In 2015, more than 10% of Malaysian patients receiving dialysis were placed on PD, of whom the majority were on CAPD (83.6%).1
Ganter first published a trial of PD for uraemia in 1923.2 Since then, PD techniques had improved considerably. Intermittent PD with high-flow and large-volume was introduced in the 1960s but failed to gain popularity among the dialysis community.3 Then, the principles of the CAPD were first described by Popovich et al where glass bottles were used to hold the dialysate.4 Two years later, Oreopoulos et al 5 proposed to replace the glass bottle with plastic bags to reduce peritonitis rate.5 A further refinement to CAPD was achieved with the introduction of the double-bag technique with a Y-disconnector in the 1980s.6
Peritonitis was a significant problem during the ‘bottle’ era where the rate was reported to be as frequent as one incident in every 3–4 patient-month. The replacement with plastic bags drove the frequency down to one episode every 9–12 patient-month but such rate still presented significant clinical problems.7 With the advent of the Y-disconnector double-bag system, peritonitis rate was lowered to approximately one episode in every 20 patient-month.3 The rate continued to improve with the later systems. A systematic review of randomised controlled trials published in 2001 indicated that peritonitis rate ranged from 1 episode in 24.8 months to 1 episode in 46.4 months.8
Malaysian studies showed marked differences in peritonitis rate between two Y-disconnect systems. In 1999, the Carex system (B. Braun Carex, Mirandola, Italy) was introduced into Malaysia in addition to the existing Ultra disconnect system by Baxter (Baxter Healthcare, Tokyo, Japan). In the ensuing equivalence study, it was found that patients using the Carex system had a 2.2 times higher risk to develop peritonitis (Incident Rate Ratio, IRR 2.18; 95% CI 1.51 to 3.14).9 Similarly, the following study that compared the ANDY-Disc system (Fresenius Medical Care [FMC], Bad Homburg, Germany) with the ultra disconnect system also revealed a 1.5 times higher risk of peritonitis, although the 95% CI overlapped the predetermined ±20% margin of equivalence. It was postulated that there was a cause-and-effect relationship between defects of the ANDY-Disc system with peritonitis.10
FMC had recently registered the Stay Safe Link system in Malaysia in October 2016. It was intended for the Stay Safe Link system to replace the existing Stay Safe system which was produced in Germany. The Stay Safe Link system will be produced in Malaysia. Both systems are similar in design apart from the use of polyvinyl chloride instead of Biofine material for the transfer tubing in the Stay Safe Link system, and slight differences in dimensions of the dialysate and drainage bags.
The rate of peritonitis was not routinely used as the assessment criteria when changing patients with ESRD from one Y-disconnect system to another. However, our experience indicates that peritonitis is a critical safety component and should always be included in the risk–benefit consideration. Thus, we intend to undertake a non-inferiority trial to determine the clinical effectiveness and safety of Stay Safe Link compared with Stay Safe.
Research hypothesis
Stay Safe Link is non-inferior to Stay Safe in terms of clinical performance and safety in patients with end-stage kidney disease on CAPD.
Study objectives
General objective
To determine the clinical performance and safety of Stay Safe Link in patients with ESRD compared with Stay Safe.
Specific objectives
Evaluation of the clinical performance of Stay Safe Link compared with Stay Safe with regards to:
Clinical effectiveness:
Delivered dialysis dose, that is, weekly Kt/V, creatinine clearance (CrCl).
Ultrafiltration volume.
Device deficiencies:
Defects and leakage of PD bag, tubing system or disc organiser.
Bacterial or other contamination of dialysate or any component of the PD system.
Evaluation of the following safety parameters
Peritonitis.
Adverse device effects (ADEs).
Serious adverse events (SAEs).
Serious ADEs (SADEs).
Unexpected SADE (USADE).
Technique failure (transfer to haemodialysis >30 days).
Trial design
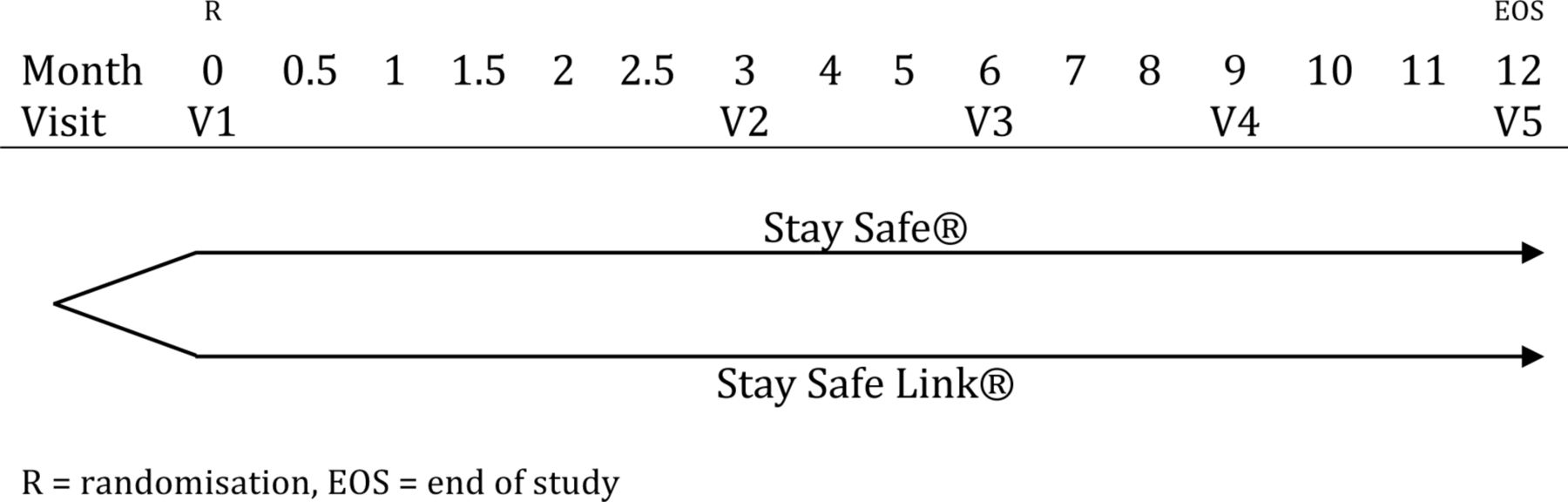
The CAPD-3 trial is designed as an open-label, parallel group, randomised controlled trial comparing Stay Safe Link with Stay Safe PD system in patients with ESRD with 1:1 allocation ratio. The trial has been registered in a public trial registry (ClinicalTrials.gov). Further trial information is available elsewhere (see online supplementary material 1). A diagrammatic representation of the trial design can be found in figure 1.
Supplemental material
{kind=link}
The study design of the CAPD-3 trial, which is an open-label randomised controlled trial. A total of 434 subjects will be equally randomised into either one of two intervention arms at the baseline visit (V1). Subsequent study visits will be conducted at an interval of 3 months (V2–V5). The 12th-month visit (V5) will also serve as the EOS visit. CAPD, continuous ambulatory peritoneal dialysis.
Methods: participants, interventions, outcomes
Study setting
Patient and public involvement
Trial patients and the public were not involved in the development of the research question or the design of the trial.
Selection of study sites
Study sites from across the country will be selected to provide a good representation of the actual CAPD patient population.
Additional consideration will be given to select study sites that have experienced nephrologists who are trained to perform the CAPD procedure. This is to minimise the influence of clinicians on operator-dependent confounding factors such as the Tenckhoff catheter survival time.
Our final selection of study site comprises 19 hospitals situated in 12 (out of a total of 13) Malaysian states. All study sites have at least one nephrologist who is familiar with the CAPD procedure and is a qualified clinical researcher.
Eligibility criteria
Inclusion criteria
18 years of age or above.
End-stage kidney failure receiving CAPD for at least 4 weeks.
Written informed consent.
Exclusion criteria
Requirement for 2.5 L exchanges.
Requirement for Stay Safe Balance.
PD-related infection (peritonitis, exit site or tunnel tract infection) in the preceding 8 weeks or during conversion.
Malfunctioning of PD catheter defined as any mechanical failures that obstruct the normal inflow and/or outflow of dialysate.11
Planned transfer to automated PD, haemodialysis or transplant within 90 days.
Pregnancy.
Any condition that compromises the integrity of the abdominal wall, abdominal surface or intra-abdominal cavity, such as documented loss of peritoneal function.
History of active alcohol or substance abuse in the previous 6 months.
Other medical condition which, in the investigator’s judgement, may be associated with increased risk to the subject or may interfere with study assessments or outcomes.
Intervention treatment
Study intervention and comparator
Eligible patients will be randomised in equal proportion between the intervention (Stay Safe Link) and the comparator (Stay Safe). The peritoneal dialysate volume to be administered each time is 2 L for all patients, but the daily frequency of administration will depend on the patient condition and treating nephrologists’ decision.
Both the intervention and the comparator are Y-disconnect systems with a preattached PD dialysate bag containing the dialysis fluid, transfer tubing, drainage bag and disc organiser to control the flow of the dialysate. The dialysate composition of both systems is identical. The system specification of Stay Safe and Stay Safe Link are listed in table 1 below:
Dialysate composition of Stay Safe and Stay Safe Link
Investigational product supply and handling
Both the interventional and comparator products are manufactured by FMC for commercial use and comply with regulatory requirements for packaging and labelling. The information to be provided to the study teams includes the manufacturer’s identification, registration number with the Malaysian National Pharmaceutical Regulatory Agency, the product manufacturing lot number and expiry date, and the chemical contents of PD fluid for both products.
PD products will be delivered directly from the manufacturer to the study subjects’ home after randomisation, and the manufacturer will provide an inventory of supplies to the study team for investigational product (IP) accountability. Any damaged or otherwise unusable IPs will be documented in the study files and notified to the sponsor and study team. At each clinic visit and phone call assessment, study coordinators will communicate with each subject to ensure the IP is supplied correctly and in sufficient quantity.
The study teams will be informed of the appropriate storage condition of the IPs. The required condition for storage is also printed on the package leaflet and the outer packaging of each carton. It is the study teams’ responsibility to inform and train study subjects on the appropriate storage conditions and handling of IPs.
Study investigators will maintain an IP accountability log throughout the study period. All study subjects will also be requested to keep an adequate ‘patient diary’ that records the frequency of their daily PD sessions, the type of dialysate used, ultrafiltration volume, weight and blood pressure. Such records will be made available to study investigators during clinic visits.
Modifications to allocated intervention
Any discontinuation or modifications to the study subject’s treatment will be at the study investigators’ discretion. Investigators are allowed to increase the frequency of the daily PD dialysis if necessary, depending on the subject’s ultrafiltration volume. However, no change to the volume of dialysate (2 L for each cycle) will be allowed. The change will be recorded in the case record form (CRF).
Any other interruptions and the duration of such interruptions to the PD treatment will be recorded accordingly.
Adherence to intervention protocol
Study subjects’ adherence to the CAPD treatment regimen is vital to ensure both study groups receive equal exposure to the IPs. This allows accurate interpretation of trial results with sufficient power to support the subsequent acceptance or rejection of the study hypothesis.
To ensure the validity of the study data, a two-tiered approach will be adopted to ensure subjects adhere to the CAPD treatment. The study coordinators will call all patients via the telephone on a 2 weekly basis (for the first 3 months) followed by monthly phone calls (for the subsequent follow-up period) to inquire if the subject experience any product defects or logistics issues that will negatively impact their treatment. Additionally, all subjects will be trained to maintain a product usage log (patient diary) as a routine practice. The proper maintenance of such log will be reinforced by the study investigators on recruitment and will be inspected by study coordinators during study visits.
Study coordinators will be instructed to examine the actual number of IP received and used by the subjects during each 3 monthly clinic visits. The details will be cross-referenced with the inventory provided by the manufacturer.
Outcomes
Assessment of clinical effectiveness
The clinical performance of the interventional product, Stay Safe Link will be evaluated to determine if the performance indicators delivered are as prescribed by the investigators. The following performance indicators will be collected:
Dialysis dose delivered, that is, weekly Kt/V and CrCl.
Ultrafiltration volume.
Defects and leakage of PD bag, tubing system or organiser.
Bacterial or other contamination of dialysate or any component of the PD system.
Assessment of safety
Evaluation of the following safety parameters will be conducted
Peritonitis.
Device deficiencies.
ADE.
SADE.
USADE.
SAE.
Technique failure (defined as the transfer to haemodialysis for more than 30 days).
The primary safety outcome of the study is the peritonitis rate. Peritonitis in this study is defined as the presence of at least 2 of the following:
Abdominal pain or tenderness or cloudy fluid.
Presence of white blood cells in the peritoneal effluent in excess of 100 cells/µL comprising at least 50% polymorphs.
Positive dialysate culture result.
All episode of peritonitis will be subjected to adjudication by two independent nephrologists. Only adjudicated peritonitis will be considered for determination of peritonitis rate. Relapsing episodes of peritonitis will not be counted separately; recurrent and repeat episodes will be counted as independent peritonitis. The terminology and definition of recurrent peritonitis, relapsing peritonitis, repeat peritonitis, refractory peritonitis and catheter-related peritonitis are in accordance with the 2016 International Society for Peritoneal Dialysis (ISPD) guidelines and Recommendations (see online supplementary material 1).12
A SADE event is any SAE caused by, or associated with, the use of the device. An SAE was defined as any adverse events that fulfil at least one of the following:
Results in death.
Is life threatening.
Results in subject hospitalisation (refers to unplanned, overnight hospitalisation) or prolongation of existing hospitalisation.
Results in a significant or persistent disability/incapacity defined as any event that results in substantial and/or permanent disruption of the subject’s ability to carry out normal life functions.
Device deficiency (DD) is defined as the inadequacy of a medical device related to its identity, quality, durability, reliability, safety or performance, such as malfunction, misuse or use error and inadequate labelling.
DD in this study includes the following:
Leakage of the dialysate bag, tubing, disc organiser or effluent bag.
Discoloured dialysate bag, tubing, disc organiser or effluent bag.
Discoloured or cloudy dialysate.
Particulate matter in dialysate.
Presence of foreign body.
Persistent kinking in the tubing which affects flow.
Malfunction of the disc device.
A DD will be classified as follows:
Device failure: A device is considered to have failed if it does not perform according to its labelling or negatively impacts the treatment while used in accordance with its labelling.
Device malfunction: A device malfunction is an unexpected change regarding the device that is contradictory to the labelling, which may or may not affect device performance.
Device misuse: A misused device (one that is used by the investigator in a manner that is contradictory to the product labelling) will not be considered a malfunction.
Patients will be instructed not to use the product if the above device deficiencies are noted before use. If the patients have used the product with descriptions consistent with those listed in (A), (B), (C), (D) or (E) as above, they will be instructed to contact the investigator immediately as they may require antibiotic prophylaxis.
Patients are required to enter into their diary the nature of the deficiencies, date of the occurrence, the quantity and lot number(s). If possible, the patients should return the product to the investigators if the product has not been used or obtain photo documentation of the deficiencies and the lot numbers.
All DD should be reported within five working days.
Participants timeline
Subjects will be assessed for eligibility to enter the study according to the inclusion and exclusion criteria. Written informed consent from the subjects will be obtained prior to any trial-related procedure.
At the baseline visit, the following data will be collected:
Subject’s demography: date of birth/age, sex, race, education level.
Background and medical history which includes any diagnosed medical conditions within the previous 6 months, history of medication and medical procedures within the previous 30 days of entry into the study.
Physical examination: vital signs (blood pressure, pulse rate), heart, lungs, fluid status.
Body weight (kg), measured with no shoes in light clothing.
Height (cm), measured with no shoes.
Laboratory evaluations
Full blood count, renal profile, liver function test, calcium, phosphate, fasting blood glucose
Fasting lipids, ferritin, transferrin saturation, weekly Kt/V, weekly CrCl.
Peritoneal equilibration test.
Dialysis treatment and care including assistance required to perform PD exchanges, prior antimicrobial exit site treatment, history of nasal Staphylococcus aureus swab and treatment.
All subjects will be required to undergo a training of at least 2 PD exchange procedures regardless of the treatment arm allocation, and the training will be completed as soon as possible following randomisation to minimise drop-outs. If the subject suffers peritonitis at training, they will be withdrawn from the trial and rescreened no earlier than 8 weeks after the resolution of that peritonitis episode.
Subsequent clinic visits will be scheduled at 3, 6, 9 and 12 months after the baseline visit. Phone call assessments will be conducted at 0.5, 1, 1.5, 2, 2.5, 4, 5, 7, 8, 10 and 11 months to evaluate subject’s progress and to remind them to complete their PD logbooks and to report any product deficiency.
At every data collection point, all subjects will be accounted for. If any subject drops out or withdraws from the study, the reason for such withdrawal (death, renal transplant, switch to haemodialysis, withdrawn consent or other reasons) will be recorded. If the subject switches to a different treatment, that is, not continuing on either arm of the study, the information of the new modality of treatment, the date of the switch and the reason for such switch will be recorded.
The study visit and procedure are outlined in table 2. Any deviation from the study procedure will be recorded in the CRF and the investigators will be notified. Protocol deviations will be notified to the Institutional Review Board (IRB).
Summary of study visits schedule and procedures
Subject participation in the study may be terminated at the investigator’s discretion. They are also free to withdraw from the study at any time for any reason. Should a subject withdraw or is withdrawn, every effort will be made to complete and report the observation as thoroughly as possible. Withdrawn or drop-out subjects will not be replaced.
Sample size and power consideration
With the reference peritonitis rate of 0.028 episodes per month and the significance level (alpha) of 0.05 using a one-sided test, 184 patients will need to be studied in each group to achieve an 80% power to detect an increase in rate ratio of at least 1.5 times, observed over a period of 12 months. Accounting for a 15% drop-out, 217 patients will be needed in each group.
Recruitment
As all study sites fall under the jurisdiction of the Ministry of Health (MOH), we recognise that all of them have similar organisational structure and working procedures to screen and select eligible subjects.
No specific advertisement will be used to improve recruitment rate. All subjects are expected to be recruited from the existing patient pool to ensure a balanced representation of the screened population.
Assignment of interventions
Allocation
Randomisation
The randomisation list will be generated using Stata Statistical Software, Version 11 (StataCorp, College Station, TX, USA) by one study personnel whose sole responsibility is to generate such lists. Permuted-block randomisation with varying block sizes will be used to generate the randomisation code. Separate randomisation codes stratified by a history of peritonitis will be generated for each centre. The block size will be concealed until the final analyses can take place.
Allocation concealment
Randomisation will be performed by three study coordinators who are independent of all other study conduct and have no interaction with the study teams. The randomisation code will be kept separately from the study site and the treatment allocation will only be released to the study investigators on a per-subject basis after the subject has been recruited into the trial.
Implementation
All subject who has given consent for participation and fulfil the study inclusion criteria will be randomised to either Stay Safe or Stay Safe Link. Randomisation will be requested by either the investigators or study coordinators via telephone calls to the three specific study coordinators who are in-charge of randomisation.
The treatment allocation will be first verbally communicated via the telephone and followed by an email that records the randomisation number, IP allocation and the unique subject ID.
Blinding
The study is an open-label trial where the investigators and study subjects know which IP they will be assigned to. However, the analysts and statisticians will be blinded to these allocations during the final analyses.
Subject retention strategy
The study visits are scheduled to conform to the routine clinical visits to minimise additional trips to the hospital. Moreover, study coordinators will be tasked to maintain frequent communication with all subjects via 2 weekly or monthly phone calls to promote adherence to the study protocol.
Statistical analysis
Baseline comparability
Demographic and baseline characteristic including age, gender, educational attainment, disease status, comorbidity and laboratory evaluations will be summarised and tabulated.
Continuous variables will be summarised by descriptive statistics, which comprise mean, median, SD, minimum and maximum. Discrete variables will be summarised by frequencies and percentages (contingency tables).
Efficacy analysis
Effectiveness will be analysed among patients with at least one measurement of delivered dialysis dose.
Safety analysis
Safety analyses will be performed on all patients who have received at least one dose of study treatment. Both per-protocol and intention-to-treat analyses (ITT) will be performed but the primary analysis will be ITT. Poisson regression will be used to compare the peritonitis rate between the CAPD systems.
Handling of missing, unused or spurious data
‘Last observation carried forward’ (LOCF) imputation technique will be performed for the demographic and baseline characteristics data. For missing efficacy data, they will be imputed by using LOCF method but no baseline data will be carried forward (in other words, a patient without any postbaseline data will be excluded from the efficacy analysis of the adequacy of dialysis).
Outliers will be identified via edit check process prior to database lock, and they will then be verified against the source document; actions with the sponsor will be defined as needed.
Data management
An appropriate data management strategy will be implemented to uphold the quality of data collected. Training on trial-related procedure will be given to investigators and study coordinators prior to the start of the study, and when the need arises thereafter.
Data management tools
This project relies on the electronic CRF (eCRF) as the main source of data collection method. In addition to the eCRF, a hard copy CRF (hCRF) will be maintained by all study sites as the secondary, backup data collection tool. Both the eCRF and hCRF are identical to each other.
The eCRF platform is Research Electronic Data Capture (REDCap) tools (V.7.1.0)13 that is hosted at Clinical Research Centre Hospital Pulau Pinang (CRC HPP). The platform’s physical server is located within the compound of CRC HPP in Malaysia, within a secure room with limited access privilege. The REDCap platform is managed by two system administrators who are the permanent staff of CRC HPP, and an information technology (IT) support personnel for hardware support.
CRF designs
The design of the hCRF is completed by members of the steering committee on which the eCRF is based. This is to ease data entry, facilitate workflow and ensure data accuracy between the two methods.
Each study personnel will be given a REDCap username and password, and can only access data from their respective sites. However, specific user privileges will be allocated to the principal investigator and data manager that allows the complete overview of study data. An audit trail will be preserved by the REDCap system to ensure data accountability.
Free text or ‘string’ data is minimised and limited only to relevant sections. Data, that is, ‘missing’, ‘not done’ or ‘not available’ will be represented as ‘NA’ in the hCRF and ‘99999’ in the eCRF.
Data collection flow
Prior to the start of the trial, all study investigators, study coordinators and monitors will be trained on good documentation practice and appropriate data entry method using both the eCRF and hCRF.
Each data point will be entered into the source document at respective study sites, and then transcribed onto the eCRF and hCRF. The eCRF or the hCRF will not be considered as the source document.
Independent clinical trial monitors will conduct periodic trial monitoring, including verifying data from source documents and to cross-check the transcribing of data points between the hCRF and eCRF. Any discrepancies or queries will be directed towards the principal investigators and/or the specific data entry personnel. Corrections or clarification must be performed in accordance with Good Clinical Practice within a stipulated time frame.
Monitoring
Data safety monitoring board
A data safety monitoring board (DSMB) will be established to review prespecified safety data at a predetermined interval (every 6 months) to ensure subject safety is protected. All members of the DSMB are independent of the study organisers and composed of nephrologists who are familiar with clinical research and CAPD treatment.
Safety data will be provided periodically to the DSMB in strict confidence as well as any other analyses of which the board may request. In the light of safety data, the DSMB will advise the steering committee, in its view, of the following action:
The study may continue without modifications.
The study may continue with modifications as suggested by the DSMB.
The study should be stopped based on the reason provided by the DSMB.
Request more data for review.
Recommend other changes as suggested by the DSMB.
No interim analysis will be performed in this study.
Harm
The study requires timely reporting of ADEs, SAE, SADEs and USADE.
Specific stopping rules will be implemented during the trial to protect study subjects. The DSMB reserves the right to temporarily suspend subject enrolment if the following occurs:
If the rate of peritonitis in the intervention arm is three times higher within the first 6 months of the study and two times higher thereafter.
If more than 1% of the IP shows product defects.
If USADE presents an unreasonable risk to subjects.
Clinical monitoring
Independent clinical monitors will be elected to conduct study monitoring at a predefined interval in order to ensure sufficient protection over study subjects and to secure the quality and integrity of the data.
The monitors will be responsible to oversee the progress of the study to make sure it is conducted in accordance with the protocols, Good Clinical Practice guideline and any applicable ethical and regulatory requirement. All clinical monitors will be trained prior to the commencement of their work and will follow a predetermined monitoring plan. All communication, including monitoring visits, phone calls and letters, between the clinical monitors and study investigators or coordinators, will be recorded.
Ethics and dissemination
Protocol amendments
Any major modification to the protocol that affects the study objectives, study design, subject population and study procedure will require agreement from the steering committee and formal approval from the Medical Research Ethics Committee (MREC).
Minor modifications such as administrative changes or corrections of typographical errors will be informed to the MREC, and expedited approval will be sought from the ethics committee.
All correspondence with the MREC will be filed by the principal investigator in the investigator study file.
Consent
Qualified nephrologists, who will act as study investigators, will introduce the trial to eligible patients. Patients will receive a copy of the Patient Information Sheet and Informed Consent Form, and the nephrologists will discuss all study aspects with the patients including but not limited to the nature of the study, its purpose, the treatments and the probability of random assignment to treatment groups, those aspects of the study that are experimental, the procedures involved including all invasive procedures and the discomfort they may entail, the possible risks including to an embryo, fetus or nursing infant where applicable, the reasonably expected benefits, the expected duration and the approximate number of patients involved and the patient’s responsibility.
All patient will also be informed that the participation is voluntary and he or she may withdraw from the study at any time for any reason, and that withdrawal of consent will not affect the subsequent medical treatment or relationship with the treating physician. The patient will also be notified in a timely manner if information becomes available that may be relevant to the patient’s willingness to continue participation in the study, any alternative procedures or treatment that may be available and the important potential benefits and risks of these available alternative procedures or treatments, any compensation for additional costs and/or injury caused to a subject attributable to participation in the study, financial expenses, if any, to the patient for participation in the study as well as prorated payment, if any, to the subject for participating in the study, any foreseeable circumstances and/or reasons under which the patient’s participation in the study may be terminated, and the person(s) to contact for further information regarding the study and whom to contact in the event of study-related injury.
Patients will be given sufficient time to have informed discussions with the nephrologists as well as their family members. Freely given written consent will be obtained from each patient involved in the study. If written consent is not possible, oral consent will be obtained if witnessed by a signed statement from one or more persons not involved in the study and stating why the patient is unable to sign the consent form.
If the patient and his or her parent or guardian are unable to read, the investigators will be required to explain to the patient the content of the patient information sheet and informed consent form point by point in the presence of an impartial witness. The witness will personally sign and date the consent form.
Each consented patient will receive one copy of the informed consent form and patient information sheet. The original consent form will be filed by the investigators in the investigator study file (see online supplementary material 2 for a sample of the English informed consent form and patient information sheet).
Supplemental material
Confidentiality
The study investigators will assure that the subject’s anonymity will be maintained and the confidentiality of records and documents that can identify the subject will be protected, respecting the privacy and confidentiality rules in accordance with applicable regulatory requirements.
All study subjects will be identified only by their assigned identification number and initial on all CRF and other records or documents submitted to the study team and sponsor. Investigators will maintain a patient identification list with complete identification information (name, contact number, identity card number) for each subject at respective study site of which will be kept in locked file cabinets in an area with limited access.
Study monitors and auditors from the CRC and sponsor and representatives from IEC or other regulatory agencies will be granted direct access to subject medical records and other study documents in order to verify the study procedures and data without violating the confidentiality of the subject. The subject will be informed of such matter during the informed consent discussion, and by signing the written informed consent form, the subject or his/her parent/guardian is authorising such access.
All electronic data recorded on REDCap will be identified by subject numbers and subject initial only. Access to the electronic database will be restricted to authorised study personnel and protected by passwords.
Access to data
Only the investigators involved in the writing committee will be given access to the final, cleaned data set. This committee will be responsible for writing the final study report and preparing the manuscripts for publication. The remaining investigators will only have access to data of their own sites unless they request such access in writing and is approved by the writing committee.
It is mandatory that the first publication is to based on data from all centres, analysed as stipulated in the protocol by the statistician at the trial coordinating centre and not by the investigators themselves unless formally agreed to by all other investigators and the writing committee.
Subjects personal information will not be divulged in any publication.
Ancillary and post-trial care
The patients who are enrolled in the study will be covered by the MOH indemnity arrangements. Patient protection and medical care will be provided by the study investigators and MOH in the event of trial-related injury.
Dissemination policy
Trial results and publication
The investigators involved will form a writing committee prior to commencing the study. The committee will be responsible for writing the final study report and preparing the manuscript for journal submission. In order to protect the scientific integrity of the study, the first publication will be based on data from all centres, analysed as stipulated in the protocol by the statistician at the trial coordinating centre. Thus, it is not expected that individual investigators will present data gathered from one centre or a small group of centres before the full initial publications.
Authorship policy
Authorship will be determined by mutual agreement and will comply with the International Committee of Medical Journal Editors (ICMJE) guidelines. Criteria for selection of additional authors will be agreed prior to the start of the study. As it will not be possible for all investigators to be named as authors in the primary publication, other investigators who have enrolled patients will be acknowledged as being part of the study team. Investigators who do not enrol any patients in the study will not be included in the list.
Acknowledgments
We used the SPIRIT checklist when writing our report.
References
Footnotes
Contributors LMO conceived of the study. WYM, BLG, SB, LM, CTL and LSH initiated the study design. WYM and CTL also are the main study personnel in trial conduct and implementation, while BLG, SB, LM and LSH are main study investigators. LMO provided statistical expertise in clinical trial design, WYM and CTL will conduct the primary statistical analysis when the trial completes. All authors contributed to the refinement of the study protocol and approved the final manuscript.
Funding The study is fully supported financially by the Ministry of Health Malaysia (MOH). The funding from the MOH covers meetings, central organisational costs and costs to conduct the trial at each individual site.
Disclaimer The study team does not receive any financial support from the manufacturer Fresenius Medical Care Sdn Bhd (FMC), and FMC has no influence over the study design, conduct or data analysis.
Competing interests None declared.
Ethics approval This study protocol and any other documents that requires approval from an Independent Ethics Committee (IEC), including the Patient Information Sheet, Informed Consent Form, subject requirement procedures and advertisements to be used (if any), and information on payments and compensations available to subjects (if any), has been submitted to the properly constituted IEC—the Malaysian Medical Research Ethics Committee (MREC)—and received unconditional approval prior to the commencement of the study (Reference: (16)KKM/NIHSEC/P17-745, dated 29 May 2017).
Provenance and peer review Not commissioned; externally peer reviewed.
Collaborators A study steering committee has been established to direct protocol development and study conduct. The steering committee is assisted by a project management team led by a certified project manager to supervise study progress. An independent Data Safety Monitoring Board has been established to review key safety data and reserves the right to suspend study enrolment should any of the study stopping rule is fulfilled. Detail descriptions of each committee and its members are reported elsewhere (see online supplementary material 1).
Patient consent for publication Not required.