Article Text

Abstract
Introduction Emergency intubation of children with abnormal respiratory or cardiac physiology is a high-risk procedure and associated with a high incidence of adverse events including hypoxemia. Successful emergency intubation is dependent on inter-related patient and operator factors. Preoxygenation has been used to maximise oxygen reserves in the patient and to prolong the safe apnoeic time during the intubation phase. Transnasal Humidified Rapid Insufflation Ventilatory Exchange (THRIVE) prolongs the safe apnoeic window for a safe intubation during elective intubation. We designed a clinical trial to test the hypothesis that THRIVE reduces the frequency of adverse and hypoxemic events during emergency intubation in children and to test the hypothesis that this treatment is cost-effective compared with standard care.
Methods and analysis The Kids THRIVE trial is a multicentre randomised controlled trial performed in participating emergency departments and paediatric intensive care units. 960 infants and children aged 0–16 years requiring emergency intubation for all reasons will be enrolled and allocated to THRIVE or control in a 1:1 allocation with stratification by site, age (<1, 1–7 and >7 years) and operator (junior and senior). Children allocated to THRIVE will receive weight appropriate transnasal flow rates with 100% oxygen, whereas children in the control arm will not receive any transnasal oxygen insufflation. The primary outcomes are defined as follows: (1) hypoxemic event during the intubation phase defined as SpO2 <90% (patient-dependent variable) and (2) first intubation attempt success without hypoxemia (operator-dependent variable). Analyses will be conducted on an intention-to-treat basis.
Ethics and dissemination Ethics approval for the protocol and consent process has been obtained (HREC/16/QRCH/81). The trial has been actively recruiting since May 2017. The study findings will be submitted for publication in a peer-reviewed journal.
Trial registration number ACTRN12617000147381.
- intubation
- apnoeic oxygenation
- airway management
- paediatrics
- nasal high-flow
- transnasal humidified rapid insufflation ventilatory exchange
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- intubation
- apnoeic oxygenation
- airway management
- paediatrics
- nasal high-flow
- transnasal humidified rapid insufflation ventilatory exchange
Strengths and limitations of this study
This is the first study investigating the use of Transnasal H umidified R apid Insufflation V entilatory E xchange in paediatric patients in the emergency and intensive care setting.
Data collected during a stressful and often unplanned procedure are validated against a video recording to ensure data accuracy and fidelity. Where there is a discrepancy between the two values, video data will be used for analysis.
Apart from the application of apnoeic oxygenation, all other aspects of the intubation process remain at clinician preference, ensuring that bias and confounding are minimised and any observed effect can be more reliably attributed to the intervention applied.
Blinding of the intervention is not possible due to visual differences in the intervention arm.
Seniority and experience of the intubating clinician may influence the likelihood of intubation success. To limit this potential confounder, there is a stratification into junior and senior operators with a preplanned subgroup analysis.
Background
Emergency intubation of the trachea in critically ill children represents one of the most challenging procedures that a clinician working with acutely ill children performs. The intubation of children with unstable physiology in the emergency department (ED) or intensive care unit (ICU) is associated with a high rate (up to 40%) of life-threatening adverse events (AEs).1 Successful emergency intubation is dependent on inter-related patient and operator factors. While a large body of literature defines optimal practice in intubation and difficult airway management in controlled theatre settings, emergency intubations in critically ill children are characterised by a deteriorating, unstable patient, sometimes with features of a difficult airway, yet are predominantly managed by ED and ICU teams rather than anaesthetists.
The incidence of paediatric emergency intubations is relatively low even in high-volume tertiary paediatric EDs, resulting in infrequent exposure of medical staff to this vital and time-critical procedure and limited practical experience in comparison to anaesthetists in an operating theatre setting. These challenges are even greater in regional and district hospitals, where paediatric emergency intubations represent relatively rare events. Therefore, techniques to optimise intubation conditions and improve patient-related and operator-dependent factors are essential to increase the safety of emergency paediatric intubations in all settings.2
The most critical phase of intubation is the period when clinicians attempt to secure the airway with an endotracheal tube (ETT); during this phase, the child is apnoeic and residual oxygen capacity of the lung is being depleted. To improve child-related factors, preoxygenation prior to induction of anaesthesia has been described since 1955 as a mechanism for maximising oxygen reserves and therefore prolonging the safe apnoeic time available for intubation particularly in emergency and difficult intubations.3 Infants and children have, in comparison to adults, a much lower tolerance for emergency intubation.4 As a result, they are more likely to experience alveolar de-recruitment and significant oxygen desaturation. Recent studies have shown that any failed attempt of emergency intubation is associated with a prolonged need for mechanical ventilation in intensive care.5 6 Preoxygenation does not supply an ongoing gas exchange and therefore there is an urgent need for newer methods to continue improved oxygenation during the apnoeic phase.
We have recently shown that the safe apnoeic time in healthy infants and children undergoing elective intubation can be substantially extended with a new technique, called Transnasal Humidified Rapid Insufflation Ventilatory Exchange (THRIVE).7 THRIVE provides high-flow humidified oxygen through nasal cannulae and allows continued peri-laryngoscopy oxygen delivery during apnoea. The benefit of THRIVE in allowing more time for safe intubation is thus likely beneficial in those difficult anatomical airways and cardiorespiratory compromise during emergency intubation. There are currently no published randomised studies investigating the use of THRIVE or other forms of apnoeic oxygenation in emergency intubation in children. In the adult literature, there have been a number of systematic reviews and meta-analyses on the topic of apnoeic oxygenation.8–13 In these reviews, there is significant heterogeneity in the studies included, and a paucity of adequately powered randomised trials. Overall, in adults there is a reduced frequency of oxygen desaturation when apnoeic oxygenation is used during intubation; however, this effect has not been demonstrated in patients with respiratory failure.
In order to improve emergency intubation conditions in children, our aim is to investigate the efficacy of THRIVE in critically ill children aged between 0 and 16 years in a large multicentre trial. We hypothesise that THRIVE reduces the frequency of life-threatening oxygen desaturation and increases frequency of first attempt success without hypoxemia in emergent intubation of children compared with standard practice. We also aim to demonstrate that this leads to a reduced proportion of AEs and reduced length of mechanical ventilation or length of stay in intensive care. This project will assess health economic impacts and cost-effectiveness of the intervention, taking into account the heterogeneity of service users, health system, geographical and economic conditions and end implications for resource allocation from the payer’s perspective.
Methods
Study design and setting
This study is a multicentre, non-blinded, randomised controlled trial evaluating the efficacy of THRIVE used for apnoeic oxygenation during emergency intubation of children aged 0–16 years. The study will be conducted in the EDs and paediatric intensive care units (PICUs) of participating hospitals.
Participants
Infants and children will be identified and recruited by treating clinicians in the ED and PICU of the participating hospitals. All patients being intubated in these locations will be screened for inclusion in the study. Patients meeting all inclusion criteria and no exclusion criteria (table 1) are eligible for randomisation.
Inclusion and exclusion criteria
In all participating centres, prospective consent will be obtained from the parent or guardian where possible. When prospective consent is not possible or practical, and local legislation allows, patients will be randomised to the study and written informed consent to remain in the study will be sought from parents and guardians at the earliest possible time after emergency stabilisation of the child (consent-to-continue). Data for children whose parents and guardians do not wish for their child to remain in the study will be handled according to local hospital policies, and the data will not be available for analysis.
Randomisation
A computer-based randomisation will be used to assign patients in a 1:1 ratio using variable block sizes. The allocation will be stratified by age (<1, 1–7 and >7 years), by level of the intended first operator seniority (junior and senior medical officer) and per site (hospital and ED or ICU). Sequentially numbered sealed opaque envelopes containing patient case report forms (CRFs) will be provided to sites and indicate the allocated treatment arm when opened. Randomisation will be undertaken by the enrolling clinician once the operator for the procedure has been determined by selecting the next number study pack.
Intervention
The definition of the different phases of intubation is pertinent for the precision of the data capture and accuracy of the primary outcome (figure 1).
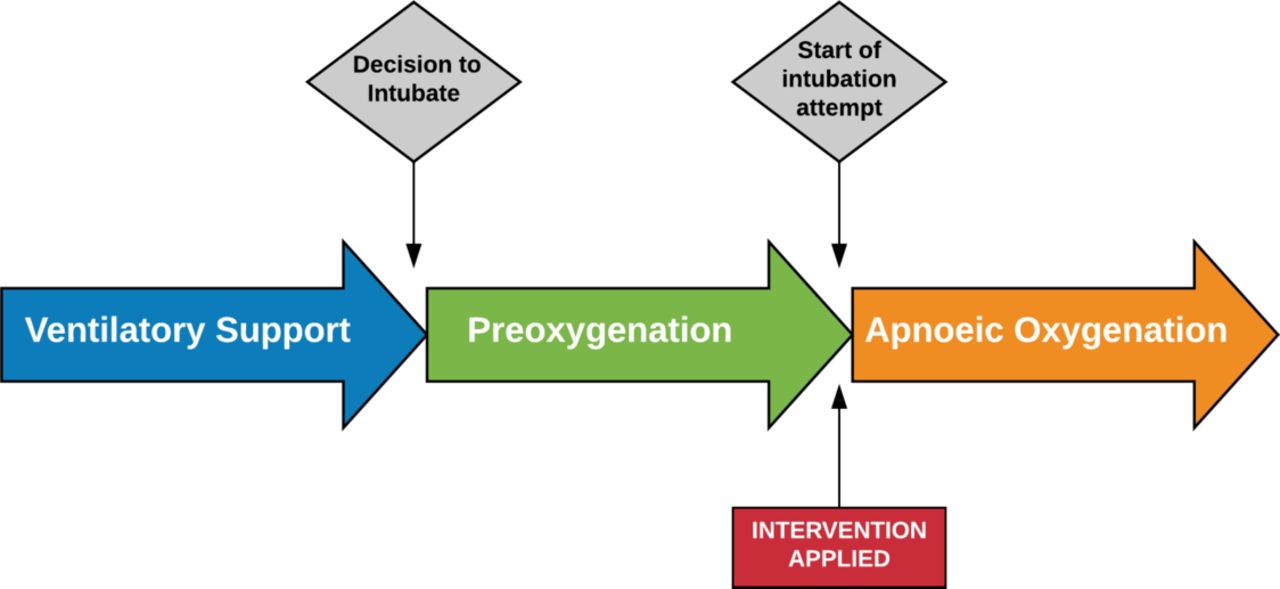
The phases of intubation are clearly defined in the protocol to ensure the intervention is applied at a standardised phase of the intubation procedure. The study allows for any method and duration of preoxygenation as per clinician standard practice, with details of preoxygenation technique and duration recorded. At the start of the intubation attempt (removal of the mask used for preoxygenation or insertion of laryngoscope blade), the intervention is applied. In the control group, all sources of apnoeic oxygen are removed at the start of intubation.
Preoxygenation phase
The preoxygenation phase is defined as the period in preparation for intubation where oxygen is delivered to the patient to maximise oxygen concentration in the functional residual capacity of the lung. For the purposes of this study, preoxygenation can be delivered by any method, including nasal high flow (NHF), at the discretion of the treating clinician. Preoxygenation should be provided with a high fraction of inspired oxygen (FiO2) for at least 3 min where possible. Where preoxygenation with high FiO2 is contraindicated or considered not appropriate for the patient by the treating clinician (eg, single ventricle physiology, unbalanced circulations), the FiO2 applied during the apnoeic phase should be the same as the FiO2 used during preoxygenation. The method used, FiO2 and duration of preoxygenation will be collected and reported.
Apnoeic oxygenation phase
The apnoeic phase begins at onset of muscle paralysis for rapid sequence induction leading to apnoea, or at the time when preoxygenation and/or non-invasive ventilation are stopped for the purpose of inserting the ETT. For the purposes of this study, the apnoeic phase will be defined as commencing at the time that the mask (or nasal prongs) used for preoxygenation or pre-intubation non-invasive ventilation (eg, by bag and mask) is removed from the face. This correlates approximately with the time of onset of paralysis or the initiation of the intubation attempt if assisted ventilation is required prior to intubation. The study intervention will be applied at the onset of this period.
Treatment arms
Patients will be randomised to receive either of the following:
Control: No apnoeic oxygenation or
THRIVE: apnoeic oxygenation with NHF.
Control
Children randomised to the standard care treatment arm will be intubated as per site-specific procedures/guidelines at clinician preference. At the onset of the apnoeic phase, as defined above, all oxygen sources must be removed from the patient during the intubation attempted.
THRIVE intervention
Patients in the THRIVE treatment arm will be provided NHF oxygen rates as per table 2, with an FiO2 of 1.0 using an Optiflow THRIVE system (Fisher and Paykel Healthcare, Auckland, New Zealand). The flow rates selected are consistent with previously published flow rates and have demonstrated an increase in safe apnoea time in children undergoing elective intubation.7
THRIVE flow rate regimen
Where preoxygenation with 100% oxygenation is contraindicated or considered not appropriate for the patient by the treating clinician (eg, single ventricle physiology, unbalanced circulations), the FiO2 applied during the apnoeic phase should be the same as the FiO2 used during preoxygenation. THRIVE is to be applied immediately when the mask for preoxygenation is removed from the face and will be maintained throughout the apnoeic period and during intubation attempts. Where NHF is used as a preoxygenation technique, it can remain in place for the intervention group, ensuring that the FiO2 is 100%, or the FiO2 is documented on the CRF in cases where high oxygen concentrations are contraindicated. THRIVE nasal prongs may be removed if rescue bag mask ventilation is required due to desaturation. The prongs will be removed on confirmation of successful ETT.
Study outcomes
The clinically relevant and patient-centred outcome measures for intubation are hypoxemia and the number of attempts for a successful intubation, both of which are strongly interlinked.14 15 Therefore, the primary outcomes for this study are defined as follows:
hypoxemic event (patient-dependent variable) and
successful first attempt intubation (operator-dependent variable).
Hypoxemia is defined as transcutaneous oxygen saturations (SpO2) of ≤90% or a SpO2 saturation difference ≥10% for patients with cyanotic congenital heart disease with known substantial right-to-left shunts measured with the bedside monitor and with an accurate quality of the trace within the period from first insertion of laryngoscope until 2 min after successful and final ETT placement (figure 2).14 16

{kind=link}
{kind=link}
In this study, the intubation period is defined as the start of intubation from removal of the mask used for preoxygenation and/or insertion of the laryngoscope blade and ends with successful intubation or abandonment of the attempt. Data collection continues for 5 min after intubation. The period from the start in intubation until 2 min after intubation is defined as the peri-intubation period and the period from 2 to 5 min defined as the post-intubation period. ETT, endotracheal tube.
A successful first attempt intubation is defined as a successful intubation at first attempt without any hypoxemia (SpO2 ≤90% or saturation difference ≥10% for right-to-left shunt). An unsuccessful intubation is either a successful first attempt intubation associated with hypoxemia or requirement for any multiple (>1) intubation attempts.
Intubation attempt is defined as a single advanced airway manoeuvre beginning with the insertion of the laryngoscope into the child’s mouth and ending when the laryngoscope is removed from the child’s mouth,17 or where there is a change in operator during the procedure even if the device is not removed.
Intubation attempt with rescue oxygenation is defined as a period with at least one unsuccessful intubation attempt followed by rescue positive pressure bag mask ventilation.
Secondary outcomes
(1) Number of intubation attempts and intubation attempts with rescue oxygenation, (2) lowest oxygen saturations during each attempt and throughout total intubation period, (3) length of mechanical ventilation, (4) ventilation free days (VFD, defined as the duration of respiratory support for all episodes with an ETT in situ for the first 28 days after randomisation censored at 28 days); VFD will be recorded as 0 in patients that died within 28 days after randomisation, (5) length of ICU stay in days, (6) length of hospital stay in days, (7) occurrence of minor AEs defined as one of the following in the period starting at the commencement of the intubation attempt until 2 min after intubation: bradycardia, not requiring treatment; hypotension, not requiring treatment; main stem bronchial intubation; oesophageal intubation with immediate recognition; emesis without aspiration; epistaxis; dental or lip trauma, (8) occurrence of major AEs (MAEs) defined as one of the following in the period starting at the commencement of the intubation attempt until 2 min after intubation: cardiac arrest with or without return of spontaneous circulation; oesophageal intubation with delayed recognition (>60 s); emesis with aspiration; hypotension requiring treatment; bradycardia requiring treatment; laryngospasm; malignant hyperthermia; pneumothorax or pneumomediastinum1 17 and (9) death defined as death during current hospital admission.
All relevant study data during the study intervention will be recorded directly onto research data form and captured with video recording of the intubation attempt and for 5-min post-ETT insertion (figure 2): (1) observations prior to first intubation attempt: this includes heart rate, oxygen saturations, blood pressure (this data is routinely recorded), (2) operator level of person performing intubation (senior: Consultant or equivalent or junior: Registrar), (3) specialty/discipline of person performing intubation, (4) intubation technique and devices used (direct, video, stylet, bougie, etc), (5) laryngeal view, grade, (6) lowest oxygen saturation during any attempt, (7) lowest blood pressure during any attempt, (8) highest and lowest pulse rate during attempt, (9) time taken for intubation attempt, (10) time of successful intubation or abandoned attempt, (11) observations immediately following successful intubation, (12) end tidal CO2 immediately after intubation (if available) and (13) minor AEs and MAEs. Additional data include (but not limited): length of mechanical ventilation, length of ICU stay and length of hospital stay.
One of the research, ED or ICU nurses will be allocated to undertake the data collection during the procedure; this will usually be the nurse allocated to be the ‘scribe’ for the procedure. In addition, a video recording of the intubation will be collected to allow validation of recorded data and collection of missing data.
Video of intubation procedure
To maximise the quality of the data collected during a stressful procedure, the entire intubation procedure will be captured by video recording. A video recording device will be provided to all investigating sites. The device will be placed in a location to have a clear view of the intubating clinician and child’s face as well as the child’s bedside monitor to ensure accurate observations are recorded during the procedure. Video will be analysed for a period of 5 min after successful intubation screening for delayed AEs. The use of the video has been extensively tested in the feasibility study and has not been perceived by nursing nor medical staff as intrusive.
Data recorded on paper CRFs will be verified against the video recording and corrections made where errors are identified by local research staff. Where there is a discrepancy between manually recorded data and video data, the video collected data will be used for analysis. Local research staff will be responsible for viewing the video and validating/collecting the required data for the CRF. Ten per cent of the videos will be centrally reviewed for accuracy of data collection. If a discrepancy is identified in the outcome measures at a site, all videos from that site will be reviewed. Video recordings of children that are consented to participate in the study constitute research records and as such will be stored on a password protected, encrypted storage device at the local site for 5 years after the study is closed and then destroyed. Video recordings of children that are not consented due to refusal or death prior to consent will form part of the child’s hospital record and will be required to be stored under the usual legal guidelines for paediatric records at that institution.
Sample size
In 2016, we audited 30 children undergoing intubation in Queensland Children’s Hospital ED, of which 12 (40%) needed a second or third attempt of intubation and 50% experienced an AE such as hypoxemia (10%), hypotension (8%), ETT misplacement (21%) and other (11%). Data obtained from 140 intubations in PICU showed similar results with 30% requiring more than one attempt. This proportion of the hypoxemia primary outcome occurrence and AEs in our own audit data is comparable to a recent publication reporting emergency intubation in the USA.1 In this setting, the authors described that in 1256 children undergoing emergency intubation, 16% of children experienced a desaturation to less than 80% during the first attempt of intubation and on average 40% required a second attempt at least. In this paper, the authors describe per age group a very similar distribution of failed attempts, with age groups defined <1, 1–7 and >7 years. A conservative estimate of the primary outcome of hypoxemia is set at 16%. We assume 90% power to detect a 50% reduction of desaturation events (hypoxia) from 16% to 8% and an alpha of 0.025 (Bonferroni’s correction for two primary outcomes), resulting in 816 participants required. For our other primary outcome, successful first attempt intubation, we estimate an increase from 60% to 80%; with 90% power and an alpha of 0.025, 258 participants are required. Therefore, an overall total sample of 960 children is required including 15% attrition which satisfies the sample size requirements for both outcomes.
Statistical analysis plan
Descriptive statistics will be utilised to report on the baseline characteristics of the total study cohort and each subgroup, as well as by site. The primary and secondary outcome measures investigating binary clinical outcomes will be compared using a χ2 test, and the difference between treatment groups will be reported as the risk difference, 95% CI and p value. For continuous outcomes, it will first be determined whether the data are normally distributed; if so, a t-test will be used for comparison, otherwise, a Mann-Whitney U test will be used. Analysis will be by intention-to-treat. Statistical significance will be set at the 0.025 level for primary outcomes, and 0.05 for other comparisons. Post-hoc power analyses may be undertaken to determine whether results found in subgroup analyses are reliable particularly for age groups (<1, 1–7 and >7 years). A preplanned secondary analysis of the outcome data will be reported for children with SpO2 <80% during intubation.
Health economics evaluation
We will undertake ex-post within-trial modelling to determine the cost-effectiveness of the intervention compared with standard care, using a cost-effectiveness approach. Unit costs will be extracted from standard sources. A standard within-trial cost utility analysis will be undertaken under the horizon of 28 days. This will compare costs and benefits in terms of resource use related to the intervention and other care and length of stay in both PICU and non-intensive care. Estimates of mean costs and CIs will be provided. Models will include sensitivity analysis. Resource use data will be collected for trial participants and the collated unit costs will be assigned to the resource utilisation to provide overall costs for both arms of the trial. The analysis will be from the healthcare provider perspective. The New Zealand country healthcare costs will be analysed separately and findings from different systems will be compared.
Ethics and dissemination
One of the primary challenges in performing research in an emergency setting is the inability to obtain true informed consent. Frequently, parents and guardians are not initially available when their child is brought into the ED. Furthermore, when parents or guardians are present, they are often too distressed by the situation to comprehend study procedures and there is not enough time to obtain informed consent.18–20
In all participating centres, prospective consent will be obtained from the parent or guardian where possible. When prospective consent is not possible or practical, and local legislation allows, patients will be randomised to the study and written informed consent to remain in the study will be sought from parents and guardians at the earliest possible time after emergency stabilisation of the child (consent-to-continue). Data for children whose parents and guardians do not wish for their child to remain in the study will be handled according to local hospital policies, and the data will not be available for analysis.
This study has ethical approval for consent-to-continue (deferred consent) for participating Australian sites. For sites in New Zealand, approval has been received for prospective consent only due to local regulatory requirements. The method of consent will be presented for all enrolled patients, along with summary data for patients eligible but not enrolled.
Results from the study will be submitted to a peer-reviewed journal for publication and for presentation at national and international conferences. Once the outcomes are known, targeted knowledge translation activities will be developed and work to incorporate the results into airway management guidelines will be undertaken.
Patient and public involvement
During the development of the protocol, parents of children being intubated were interviewed regarding the acceptability of research participation during a time-critical procedure and the acceptability of video recording during the procedure. Parents were supportive of research in these situations, and also reported no concerns regarding the use of video recording during the procedure. Participants are asked if they would like to receive a copy of the outcomes of the study during the consent process and an email address is collected to facilitate distribution of any relevant publications.
Time frame
It is anticipated that a 3-year recruitment period is needed to achieve the required sample size.
Adverse events and monitoring/reporting
The Data and Safety Monitoring Board (DSMB) consists of an anaesthetist, a paediatric intensivist and a statistician. None of the DSMB members will be involved in recruitment of study patients at their site. DSMB members will not be supervised by any study investigator, or participate as investigators in any study currently under review by this DSMB. The primary objective of the DSMB is to monitor the safety of the intervention and the validity and integrity of the data from the Kids THRIVE study. Additionally, the DSMB will evaluate the pace of recruitment and will make recommendations to the Kids THRIVE Chief Investigator(s) and Steering Board regarding the continuation, modification or termination of the study.
AE data are collected as part of the study design and form the primary and secondary outcomes of the study. Conditions that are present at screening and do not deteriorate will not be considered AEs. Patients participating in this trial are critically ill and the reason for requiring the intubation is often respiratory, neurological or cardiovascular failure with acute risk of death. Except for death during or within 1 hour of intubation and oesophageal intubation with late recognition, AEs as listed in the outcome parameters are expected and according to current clinical practice ‘accepted’ outcomes. Hence, these outcomes are not routinely reported as AEs nor to the DSMB. However, if any of the attending clinicians suspect an AE that is related to the study design, such an AE will be reported.
Serious AEs are defined as follows:
Cardiac arrest or death during or within an hour of the intervention.
Oesophageal intubation with late recognition
Any serious AE will be reported to the Human Research Ethics Committee (HREC) within 24–72 hours of occurrence, in accordance with the safety reporting policy of the HREC. Set DSMB review points on the progress and safety of the trial are predefined as after the primary outcome is known for 200 children.
Data accuracy and integrity
Intubation and ventilation of a child in the ED and ICU settings represent a highly stressful event and require significant human resources to assemble equipment while effectively managing a critically unwell patient. As a result, attention to data collection for a research study is often overlooked, with the immediate needs of the patient always taking priority. This study is dependent on high-quality and accurate data at a critical moment of the patient’s management, and the potential for missing or inaccurate data collection is high. It is also recognised that significant bias can be introduced to a study of this type when relying on retrospective recall of critical data values. A retrospective review of intubation records compared with video-recorded data has highlighted that there is a significant under-reporting of AEs, especially oxygen desaturation, and an over-reporting of first-attempt success rates, in the magnitude of 21% and 12%, respectively, when reliant on operator-/observer-reported data.21
For this reason, a video recording of the intubation procedure will be captured for data verification purposes and to protect the integrity of the primary end point. Where a discrepancy between the manually recorded data and video-recorded data exists, the video-recorded data will be used for analysis as it can be objectively confirmed.
Current status of trial
The study enrolment has commenced in May 2017 and the sites involved are as follows:
Queensland Children’s Hospital, Townsville Hospital, Gold Coast University Hospital, Royal Children’s Hospital Melbourne, Womens and Children’s Hospital Adelaide, Perth Childrens Hospital and Starship Children’s Hospital Auckland. Expansion to additional sites is planned.
Significance
This large multicentre randomised trial may help define the role of THRIVE during emergency intubation in infants and children. The intubation of a child in the emergency setting places a tremendous emotional strain on both the child and parents. Dependent on the skill level and experience of the clinician performing the procedure, the professional stress level may also be very high as intubation is a high-risk procedure, which needs to be accomplished in a timely manner. The study will provide high-fidelity data using video recording of the study interventions and the findings will easily be translated into clinical practice.
Acknowledgments
The authors would like to thank the parents and children participating in this trial and the the medical, nursing and research teams in the participating sites for their help in study setup, recruitment, data collection and monitoring of study data.
References
Footnotes
Patient consent for publication Not required.
Contributors SG, AS and SH were responsible for identifying the research question and contributing the drafting of the protocol. BGe, SE, SSG, NS, SRD, AG, KF, AC, LJS, MF, KR and TW have contributed to the development of the protocol and study design. BGa developed the health economic measures and analysis in the study. KG provided expert statistical advice and input. SG was responsible for drafting this paper, with comments and feedback from all other authors. All authors attest to having approved the final manuscript. SG and AS take responsibility for the manuscript as a whole.
Funding This research is supported by a project grant from the Emergency Medicine Foundation, a project grant from the Thrasher Research Fund and a project grant from the National Health and Medical Research Council (NHMRC). OptiFlow THRIVE™ equipment and consumables have been supplied free of charge for this study by Fisher and Paykel Healthcare, Auckland, New Zealand.
Competing interests SG, AS and SRD received travel support from Fisher and Paykel Healthcare.
Ethics approval The study protocol has been reviewed and approved by ethics committees in Australia (Children’s Health Queensland Human Research Ethics Committee, HREC/16/QRCH/81) and New Zealand (Health and Disability Ethics Committee 17/NTA/120).
Provenance and peer review Not commissioned; externally peer reviewed.
Collaborators Dr Philip Sargent, Gold Coast University Hospital, Dr Christa Bell, Gold Coast University Hospital