Article Text

Abstract
Introduction Abdominal wall hernias are a common source of morbidity and mortality. The use of biological mesh has become an important adjunct in successful abdominal wall reconstruction. There are a variety of biological mesh products available; however, there is limited evidence supporting the use of one type over another. This study aims to compare the performance (eg, the rate of hernia recurrence) of either a crosslinked biological mesh product or a non-crosslinked product in patients undergoing abdominal wall reconstruction.
Methods and analysis This is a single-centre, dual arm randomised controlled trial. Patients requiring abdominal wall reconstruction will be assessed for eligibility. Eligible patients will then undergo an informed consent process following by randomisation to either (1) crosslinked porcine dermis mesh (Permacol); or (2) non-crosslinked porcine dermis mesh (Strattice). These groups will be compared for the rate of hernia recurrence at 1 and 2 years as well as the rate of postoperative complications (eg, surgical site infections).
Ethics and dissemination This study has been approved by the institution’s research ethics board and registered with clinicaltrials.gov. All eligible participants will provide informed consent prior to randomization. The results of this study may help guide the choice of biologic mesh for this population. The results of this study will be published in peer-reviewed journals as well as national and international conferences.
Trial registration number NCT02703662.
- hernia
- biologic mesh
- hernia recurrence
- abdominal wall reconstruction
- hernia repair
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
First, clinical randomised controlled trial designed to evaluate the performance of two common types of biological mesh.
The nature of the intervention groups does not allow for blinding of the surgical team.
This is a single-centre study.
Introduction
Abdominal wall hernias are a common challenge for surgeons and represent a significant financial burden on the healthcare system.1 An increasing number of patients present with large and complex abdominal wall defects which introduce a unique set of challenges. The approach to complex abdominal wall reconstruction has evolved over the last three decades and includes techniques such as component separation, modified component separation and free tissue flap reconstruction.
Regardless of the technique used, evidence suggests that hernia recurrence rates are reduced with the use of mesh.2 3 In the presence of microbial contamination or infection, however, early evidence has suggested that there is a marked increase in hernia recurrence and mesh infection when using synthetic prostheses.4 Biological meshes (BMs) have been promoted as an option in abdominal wall reconstruction in the presence of both field contamination and infection.5–8 More recent evidence has encouraged some caution in the universal adoption of BMs for this purpose.9 10 Nonetheless, BMs remain a common adjunct in complex abdominal wall reconstruction in certain scenarios.11
A number of BMs are currently available on the market. BMs are derived from human (allograft) or animal (xenograft) dermis, pericardium or intestinal submucosa. These tissues are processed to remove any immunogenic material and, as a result, are rendered acellular. After processing, the extracellular matrix remains and is used as a scaffold by host tissues.
An important feature which differentiates BMs is the use of collagen crosslinking. The chemical crosslinking process is intended to increase the tensile strength of the mesh and render the collagen more resistant to breakdown by host tissues. However, there is also evidence that crosslinked BMs produce a more severe inflammatory reaction resulting in encapsulation of the mesh rather than integration with the host tissues.12
To date, there have been no prospective randomised controlled trials that compare various BM materials in the context of abdominal wall reconstruction.13 14 This study aims to compare the efficacy of two commonly used BMs: Strattice (LifeCell), a non-crosslinked porcine dermis; and Permacol (Covidien), a crosslinked porcine dermis. Given that there is a significant price difference between these products, the results of this study could significantly influence the preferred BM for abdominal wall reconstruction and the associated cost for the healthcare system.
Methods and analysis
Overview
The study will be a single-centre, dual-arm, parallel randomised controlled trial. Patients requiring abdominal wall reconstruction with the use of a BM will be randomised in a 1:1 ratio to Strattice (non-crosslinked, LifeCell) or Permacol (crosslinked, Covidien). We hypothesise that these porcine dermis-derived BMs will be equivalent in terms of hernia recurrence and postoperative complications. This study protocol was constructed in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 guidelines.15 A SPIRIT diagram detailing the timing of screening, randomisation, allocation and assessment is provided in figure 1.
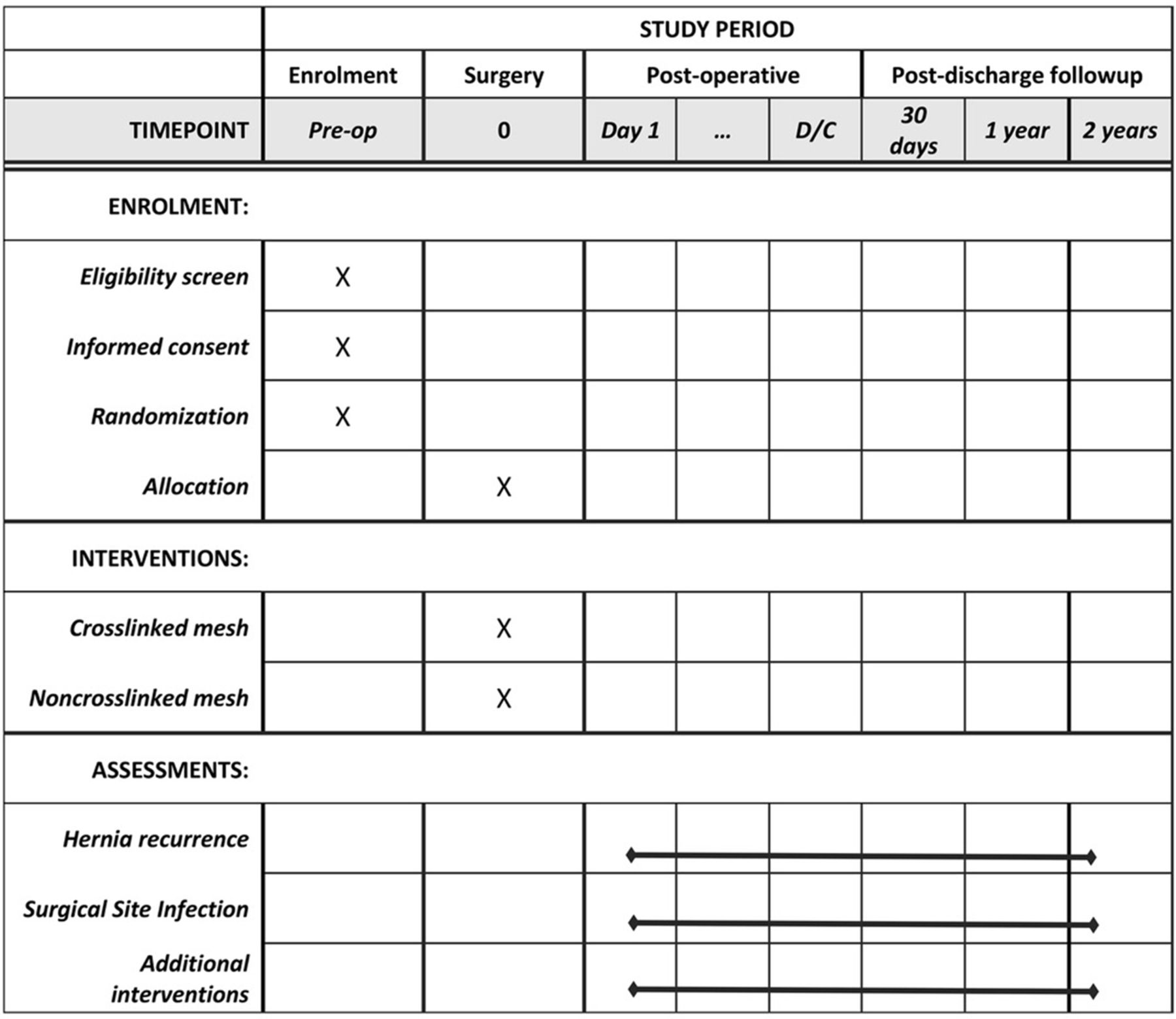
Standard Protocol Items: Recommendations for Interventional Trials diagram describing schedule of enrolment, interventions and assessments.
Trial design
Incisional hernias are a common complication following abdominal surgery. Some studies report hernia rates as high as 32%, even with the use of synthetic mesh.2 Every planned abdominal wall reconstruction much take into account patient factors (eg, age, obesity, immunosuppression and pulmonary disease) as well as technical and anatomical factors (eg, presence of contamination or infection, size and location of the defect). The trial described below includes patients from across this spectrum of real-world variability and therefore adopts a pragmatic trial design. The explanatory versus pragmatic nature of the trial is summarised visually using the Pragmatic Explanatory Continuum Indicator Summary (PRECIS-2) wheel (figure 2).16

{kind=link}
{kind=link}
Description of trial design using Pragmatic Explanatory Continuum Indicator Summary wheel.
Patient and public involvement
No patients were involved in the design of this study, nor will they be directly involved in the recruitment to or conduct of the study. Results of the study will be disseminated to patients at their request.
Abdominal wall hernias can have a significant impact on patient quality of life. Accordingly, hernia recurrence after attempted abdominal wall reconstruction represents a significant burden on this patient population. This is consistently reflected in our own experience with patients at our centre and forms the basis for the research question and chosen outcome measures.
Setting
The study will take place at the Foothills Medical Centre (FMC), a University of Calgary affiliated tertiary care hospital located in Calgary, Alberta, Canada. The FMC is a Trauma Association of Canada accredited level 1 trauma centre and high-volume surgical oncology referral centre which provides specialised surgical services for southern Alberta, southeast Saskatchewan and southwest British Columbia. Additionally, FMC serves as a referral centre for complex abdominal wall reconstruction. The two surgeons involved in this study (CGB and AWK) are fellowship-trained trauma surgeons with additional training and special interest in abdominal wall reconstructions.
Eligibility criteria
The study population will consist of adult patients, 18 years or older, undergoing an abdominal wall reconstruction. All patients eligible for abdominal wall reconstruction will routinely undergo abdominal CT for operative planning. Criteria for BM use include large abdominal defects with significant contamination or the presence of an ostomy, loss of abdominal wall secondary to trauma or necrotising infection, or abdominal wall reconstruction in patients identified as being at high risk for infection. A large incisional hernia was defined as having a minimum diameter of greater than or equal to 10 cm, in keeping with consensus definitions.17 The appropriateness of a BM will be determined via consensus between the two surgeons involved in the trial (CGB and AWK). Any study patient with a Centres for Disease Control and Prevention grade IV wound infections will be downgraded to grade III prior to intervention. Patients will be excluded if they are unable or unwilling to provide informed consent.
Consent
Eligibility will be determined by an attending surgeon following a formal outpatient assessment. Eligible patients will be contacted by a research study assistant who will provide additional information regarding the study. Final consent will then be obtained in the clinical setting by the study nurse or attending surgeon if needed (figure 1).
Randomisation and blinding
Once consent has been obtained, participants will be randomised by research staff using a block randomisation model (block size 4). Allocation will be provided to the surgeon in a concealed envelope the day prior to surgery. Patients and the data analyst will be blinded as to the allocated intervention (eg, type of BM used). Charts will be blinded for data abstraction.
Interventions
Participants will be allocated to one of two types of BM:
Crosslinked porcine dermis (Permacol; Covidien): Chemical crosslinking is performed using a proprietary process with non-calcifying hexamethylene diisocyanate. Permacol is available in sheets of a variety of sizes ranging from 1×4 to 28×40 cm.
Non-crosslinked porcine dermis (Strattice; LifeCell): Strattice is available in sheets in a variety of sizes ranging from 6×6 to 25×40 cm.
Data collection
Data will be collected from both electronic and paper-based medical records by study staff on standardised report forms. Patient and hernia characteristics will be recorded as well as details regarding the surgical technique and postoperative complications. A sample of recorded data points are included in box 1. After hospital discharge, patients will be assessed by their attending surgeon at approximately 30 days, 3, 6, 12 and 24 months. All study documents will be maintained in a secure location in a locked office.
Sample data collection
Patient characteristics
Age, sex, medical comorbidities, American Society of Anesthesiologists classification.
Preoperative chemotherapy, immunosuppression.
Preoperative albumin level.
Previous abdominal surgery.
Hernia characteristics
Length and width.
Volume of abdominal viscera outside of peritoneal domain.
Presence of bacterial colonisation or infection.
Surgery
Surgeon, technique, association procedures.
Duration of procedure, estimated blood loss, transfusion requirements.
Skin prep solution, antibiotic prophylaxis.
Wound closure technique, use of drains, dressing type.
Complications
Acute kidney injury, pneumonia, venous thromboembolism.
Surgical site infection (superficial, deep, organ space).
Need for urgent reintervention.
Outcomes
Outcomes will be assessed by the attending surgeon and other study authors. Inpatient assessments will include daily physical examinations, bloodwork and other tests as clinically indicated. The primary outcome is hernia recurrence at 1 year. Secondary outcomes include postoperative complications (eg, surgical site infection and need for reintervention) and hernia recurrence at 2 years. Hernia recurrence will be determined by the attending surgeon throughout the follow-up period. Recurrence will be confirmed with CT.
Sample size
Across studies, the reported rate of hernia recurrence following abdominal wall reconstruction with BM is 15%.18 The rate of success is therefore estimated to be 85%. Sample size for an equivalence trial was then calculated using: (1) significance level (alpha) of 0.05; (2) power (beta) of 90% and (3) an equivalence limit of 25%. Forty-five patients will be randomised to each intervention group. Historically, the follow-up rate following complex abdominal wall reconstruction at our institution is excellent with no patients having been lost to follow-up. We have therefore not accounted for this in our sample size calculation. Based on this sample size and the volume of complex abdominal wall reconstruction at the trial centre (more than 35 per year), we expect to complete recruitment within 2–3 years. Patient enrolment began on 26 October 2017.
Analysis
An intention-to-treat analysis will be used. Descriptive statistics will be used to describe the patients in both treatment arms. Continuous variables will be compared using Student’s t-test and the Mann Whitney U test. The χ2 tests or Fisher’s exact test will be used to compare categorical variables. Multivariate analysis will be done to compare hernia recurrence rates of the different managements received but also to account for other potential confounding variables. All variables with a p value <0.2 on univariate analysis will be entered into a multivariable logistic regression analysis to identify independent risk factors for hernia recurrence and morbidity. Statistical analysis will be performed with STATA.
Conclusion
BMs are established adjuncts in abdominal wall reconstruction in appropriately selected patients. This includes reconstruction in the presence of field contamination or infection. BMs vary significantly in terms of their source and processing, technical characteristics and cost. To date, no prospective, randomised controlled trials have been performed to evaluate the performance of two of the most commonly used types of BM. This trial aims to compare a crosslinked porcine dermis derived mesh (Permacol) with a non-crosslinked porcine dermis derived mesh (Strattice). There is a significant price difference between the two products (ie, the crosslinked product is approximately twice as expensive) and therefore the results of this trial could significantly influence the preferred choice of BM for abdominal wall reconstruction. Additionally, once an ideal BM has been identified, it can then be compared directly to an ideal synthetic mesh to help define the future role of BM in abdominal wall reconstruction. Given that incisional hernias are a common entity, this could have a significant impact on the healthcare costs associated with abdominal wall reconstruction.
References
Footnotes
Patient consent for publication Not required.
Contributors CGB and AWK are the principal investigators and have coordinated all the phases of trial design, statistical analysis plan and drafting of the protocol. TLE participated in study coordination. DAC critically appraised the trial design, participated in study coordination and wrote the manuscript. All authors contributed to the writing of the manuscript and agreed with submission of the final version for publication.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Ethics approval This study has received approval from the Conjoint Health Research Ethics Board at the University of Calgary (REB 15-1826). The study protocol, informed consent form and other submitted documents were reviewed and approved. Dissemination policy: final results will be publicly disseminated regardless of the study outcomes. A report releasing study results will be submitted for publication in an appropriate journal, approximately 3 months after finishing data collection. This study has been approved by the institution’s research ethics board and registered with clinicaltrials.gov. All eligible participants will provide informed consent prior to randomisation. The results of this study may help guide the choice of biological mesh for this patient population. The results of this study will be published in peer-reviewed journals as well as national and international conferences.
Provenance and peer review Not commissioned; externally peer reviewed.