Article Text

Abstract
Objectives To compare efficacy/safety of oral tramadol 75 mg/dexketoprofen 25 mg (TRAM/DKP) and TRAM 75 mg/paracetamol 650 mg (TRAM/paracetamol) in moderate to severe pain following surgical removal of impacted lower third molar.
Design Multicentre, randomised, double-blind, placebo-controlled, phase IIIb study.
Participants Healthy adult patients scheduled for surgical extraction of at least one fully/partially impacted lower third molar requiring bone manipulation. 654 patients were randomised and 653 were eligible for analysis.
Interventions Surgery was performed under local anaesthetic. No sedation was permitted. Patients rated pain intensity (PI) using an 11-Numerical Rating Scale (NRS) (0 no pain; 10 worst pain). Participants experiencing moderate/severe pain (≥4) within 4 hours of surgery were randomised (2:2:1 ratio) to a single oral dose of TRAM/DKP 75/25 mg, TRAM/paracetamol 75/650 mg or placebo.
Main outcome measures Efficacy was based patients’ electronic diaries. Analgesia and pain were recorded as follows: pain relief (PAR) on a 5-point Verbal Rating Scale (0=‘no relief’, 1=‘a little (perceptible) relief’, 2=‘some (meaningful) relief’, 3=‘lot of relief’, 4=‘complete relief’) at the predefined postdose time points t15 min, t30 min, t1 hour, t1.5 hour, t2 hour, t4 hour, t6 hour and t8 hour and PI on the 11-point NRS at t0 and at the same predefined postdose time points. Onset of analgesia documented using double stopwatch method over a 2-hour period. Primary endpoint was total pain relief over 6 hours (TOTPAR6). Rescue medication was available during the treatment period.
Results TRAM/DKP was superior to TRAM/paracetamol and placebo at the primary endpoint TOTPAR6 (p<0.0001). Mean (SD) TOTPAR6 in the TRAM/DKP group was 13 (6.97), while those in the active control and placebo groups were 9.2 (7.65) and 1.9 (3.89), respectively. Superiority of TRAM/DKP over active comparator and placebo was observed at all secondary endpoints. Incidence of adverse events was comparable between active groups.
Conclusions TRAM/DKP (75/25 mg) is effective and superior to TRAM/paracetamol (75/650 mg) in relieving moderate to severe acute pain following surgical removal of impacted lower third molar, with a faster onset of action, greater and durable analgesia, together with a favourable safety profile.
Trial registration number EudraCT 2015-004152-22 and NCT02777970.
- oral medicine
- pain management
- primary care
- public health
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The main strength of this study is that with nearly 800 patients screened, it is one of the largest to be carried out in patients with moderate to severe acute pain in the impacted third molar extraction model.
The rigorous study design—multicentre, randomised, double-blind, placebo-controlled, phase IIIb—coupled with the large patient cohort means that the quality of the evidence is high.
This is a unique head-to-head study of tramadol/dexketoprofen against tramadol/paracetamol an established combination rather than just a placebo-controlled study.
The results show that tramadol hydrochloride/dexketoprofen 75/25 mg oral fixed combination is effective and superior over tramadol hydrochloride/paracetamol 75/650 mg in relieving moderate to severe acute pain following surgical removal of impacted lower third molar.
One of the limitations of this study is as this was a single-dose study that assessed the short-term analgesic efficacy of tramadol/dexketoprofen in comparison with tramadol/paracetamol and this could be addressed in a future multidose study of longer duration.
Introduction
The majority of patients with moderate to severe acute postoperative pain report inadequate pain relief (PAR).1 This is in spite of well-known and published negative impact of inadequate PAR on clinical outcomes such as time to functional recovery, postsurgical complications, pain chronification and quality of life. It is consequently associated with increased healthcare costs.1–4 Attaining adequate PAR with monotherapy is difficult and multimodal analgesia is now accepted as the cornerstone of effective pain treatment. Combining analgesics with diverse mechanisms of action and potential synergistic effects means that a wider spectrum of pain can be covered, and lower doses of single drug components can be administered, thus enhancing efficacy and minimising adverse events (AEs).5
Dexketoprofen (DKP) is a well-known inhibitor of cyclooxygenase (COX-1 and COX-2) with proven analgesic and anti-inflammatory efficacy in a wide spectrum of acute pain syndromes; it appears to be as effective as the double dose of the racemic ketoprofen, but with a faster onset of analgesia. Its fast onset of action and favourable pharmacokinetics profile are enhanced by its formulation as trometamol salt, which has an increased bioavailability compared with the free drug. The rapid dissolution and absorption, (tmax between 0.25 and 0.75 hour) ensure rapid PAR which is crucial for the effective management of acute pain.6–8 DKP’s efficacy is complemented by its favourable safety profile with clinical data showing an AE profile similar to that of the new generation NSAIDs.8
Tramadol (TRAM), a μ-opioid receptor agonist, norepinephrine and serotonin reuptake inhibitor, is a central-acting analgesic and also shows a peripheral and local analgesic effect.9 10 Its opioid and non-opioid mechanisms are thought to act synergistically on descending inhibitory pathways in the central nervous system. TRAM’s analgesic efficacy is complemented by a long duration of action (half-life around 6 hours) and by a safety profile that favours TRAM over other opioids.8
The doses of TRAM hydrochloride 75 mg plus DKP 25 mg were selected as the optimal for a combination in a previous dose-finding study11; subsequent multiple-dose clinical trials in well-established models of acute visceral and somatic pain12 confirmed the combination of TRAM hydrochloride 75 mg with DKP 25 mg (TRAM/DKP) as an effective analgesic for the control of moderate to severe acute pain.13–16 TRAM/DKP offers a number of important advantages including: proven efficacy and tolerability with a 25% overall reduction in the opioid dosage, improved compliance, as well as a convenient mode of administration. TRAM/DKP (75/25 mg) (film-coated tablet) has been registered and released for commercial use in Europe.17
Direct comparisons of newly released drugs with older agents are rarely performed in clinical trials, although this kind of evidence is valuable to improve appropriateness of pharmacotherapy in clinical practice.18 This lack of data can make it difficult for clinicians to determine the optimal therapeutic option.19 To provide a meaningful comparison of the analgesic capabilities of TRAM/DKP fixed combination versus oral TRAM hydrochloride 75 mg/paracetamol 650 mg (TRAM/paracetamol), a head-to-head clinical trial was necessary.
The present phase IIIb trial (Dexketoprofen Analgesic eVolution wIth tramaDol) was designed to compare the analgesic efficacy and safety of a single dose of the fixed oral combination TRAM/DKP (75/25 mg) versus TRAM/paracetamol (75/650 mg) in postoperative moderate to severe pain following extraction of impacted lower third molars. The model has a proven record of assay sensitivity to compare efficacy, including onset of analgesic action, in head-to-head trials.11 20 21
Methods
It was performed at 18 centres in five European countries (Hungary, Italy, Poland, Spain and UK) and was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. The clinical phase of the study started in April 2016 and was concluded in February 2017.
Patients
Healthy adult patients (>18 years of age) scheduled to undergo surgical extraction of at least one fully or partially impacted lower third molar requiring bone manipulation were included in the trial. Criteria for randomisation included postoperative pain of moderate to severe intensity (Numerical Rating Scale, NRS ≥4) within a 4-hour time lapse after the completion of the surgery.
Pregnant or breastfeeding women or women of childbearing potential not using adequate contraception were excluded from the study. The following conditions did not permit participation in the study: known allergy or hypersensitivity to study treatments, rescue medication (RM) or any other NSAIDs except those permitted in the study protocol, opioids and acetyl salicylic acid; history of peptic ulcer, gastrointestinal disorders induced by NSAIDs or gastrointestinal bleeding; Crohn’s disease or ulcerative colitis; severe asthma; moderate to severe renal dysfunction; severe hepatic dysfunction or cardiac dysfunction; active bleeding or coagulation disorders; history of or current epilepsy; history of drug or alcohol abuse; as well as history/presence of any illness or condition that, in the opinion of the Investigator, might pose a risk to the patient or confound the efficacy and safety results of the study. Patients who had received any investigational drug or participated in any other clinical trials within the previous 4 weeks were also excluded. Patients who had taken, sedatives, hypnotic agents or analgesics within 12 hours before surgery and 8 hours postdose (or 5 days prior to the surgery day in case of COX-2 inhibitors) were not considered eligible. Moreover, subjects under chronic opioid treatment, or using and not suitable for withdrawing, within 48 hours (or five half-lives, whichever the longer presurgery), drugs posing a risk to the patient and for 24 hours postdose, were also not enrolled. Lastly, patients were excluded if any surgical complication occurred that, in the opinion of the investigator, would interfere with the study procedures or assessments.
Study design
This was a multicentre, randomised, double-blind, double-dummy, parallel-group, placebo and active-controlled, single-dose, phase IIIb study including three treatment arms. Participation in the study lasted for approximately 3 weeks for each patient and was made up of: a screening period, (within 2 weeks before randomisation), including the presurgery procedures to be completed at least 1 day prior to surgery and ending within the 4 hours qualification postsurgery; randomisation and treatment administration (day 1, t0) followed by an 8-hour assessment period during which patients recorded efficacy data using an electronic diary (eDiary) both at site (up to t2 hour) and out of site (up to t8 hour); followed by an end of study visit (6±1 days after randomisation).
Surgery was performed under local anaesthetic using 2% lidocaine (with 1:80.000 epinephrine) up to a total volume of 5.4 mL per molar. No sedation was permitted. After surgery, patients rated pain intensity (PI) using an 11-NRS ranging from 0 (no pain) to 10 (worst pain).22 23 Participants experiencing moderate to severe pain (≥4) within 4 hours after surgery were randomised with a 2:2:1 ratio to a single oral dose of TRAM/DKP 75/25 mg, TRAM/paracetamol 75/650 mg or placebo. The inclusion of a placebo group was deemed acceptable considering the self-limiting nature of postoperative dental pain, the short duration of the study, and the fact that an effective RM was provided to participants requesting additional PAR; this is in line with recent academic recommendations.24 RM consisted of oral ibuprofen (400 mg) and was permitted during the 8-hour postdose assessment period up to a maximum of two tablets at a minimum interval of 4 hours.
Randomisation and masking
Randomisation was performed through Interactive Voice/Web Response System according to a computer-generated randomisation sequence. Participants, healthcare providers, medical monitors, other personnel involved in the conduction of the trial, data collectors and biometricians were unaware of the treatment that participants were receiving. Double-blind conditions were ensured by the application of double dummy technique.
Sample size calculation
A sample size of 230 patients per active arm was considered adequate to demonstrate the non-inferiority of TRAM/DKP compared with TRAM/paracetamol assuming a non-inferiority margin of 20%, a power of 80% and an overall significance level of 2.5% (one sided). The mean total pain relief over 6 hours (TOTPAR6) for TRAM/paracetamol is assumed equal to 7.4 (SD 6.30) based on a previously published trial.25 One hundred and fifteen patients in the placebo arm were considered sufficient to demonstrate the superiority of both TRAM/paracetamol and TRAM/DKP versus placebo for model sensitivity. Assuming about 10% of major protocol violators and 20% of screening failure rate a total of 640 patients needed to be randomised and 800 patients were expected to be screened.
Efficacy evaluation
Evaluation of efficacy was based on data entered by patients in eDiary. Analgesia and pain were recorded by using the following measures: PAR on a 5-point Verbal Rating Scale (VRS) (0=‘no relief’, 1=‘a little (perceptible) relief’, 2=‘some (meaningful) relief’, 3=‘lot of relief’, 4=‘complete relief’)26 27 at the predefined postdose time points t15 min, t30 min, t1 hour, t1.5 hour, t2 hour, t4 hour, t6 hour and t8 hour and PI on the 11-point NRS23 24 28 at t0 and at the same predefined postdose time points.
The onset of analgesia was documented using the double stopwatch method over a 2-hour period postdose. Following treatment, two stopwatches were automatically activated in the eDiary. Subjects were instructed to stop the first stopwatch when they felt ‘first perceptible’ PAR (FPPAR, ie, at the moment they first felt any PAR whatsoever) and the second when they experienced a ‘meaningful’ PAR (MPAR, ie, when the relief from pain became meaningful to them). The FPPAR and MPAR could be alternatively defined by using the time point when PAR is assessed as 1 and 2, respectively, in case the stopwatch was not used or used after the recording of the PAR equal to 1 or 2. Furthermore, an overall assessment of the study medication was reported through patient global evaluation (PGE) on a 5-point VRS (1=poor, 2=fair, 3=good, 4=very good, 5=excellent) at the end of the 8-hour assessment period or immediately before the RM intake.22 The time between treatment administration and first intake of RM, and percentage of patients requiring RM were also evaluated. After first intake of RM, patients were no longer required to use the eDiary, including the stopwatch functionality.
Primary and secondary endpoints
The primary efficacy endpoint was TOTPAR, calculated as the weighted sum of the PAR scores measured according to a 5-point VRS, over 6 hours postdose (TOTPAR6). Secondary efficacy endpoints included the time course of mean PAR and PI scores over 8 hours; TOTPAR over 2, 4 and 8 hours postdose and the percentage of maximum calculated TOTPAR (% max TOTPAR) over 2, 4, 6 and 8 hours; sum of pain intensity difference (SPID) and the percentage of maximum calculated SPID (% max SPID) over 2, 4, 6 and 8 hours; percentage of responders in terms of PAR or PI reduction, namely subjects who achieved at least 50% of max TOTPAR or at least 30% of PI reduction versus baseline at prespecified time points over the 8 hours, respectively; time to FPPAR; time to confirmed FPPAR (ie, time to FPPAR if confirmed by experiencing MPAR) and time to MPAR; percentage of patients who achieved FPPAR, confirmed FPPAR and MPAR within 30 min, 1 hour and 2 hours; PGE at 8 hours or whenever the patient used RM; time of first intake of RM since study drug intake; percentage of patients using RM at 2, 4, 6 or 8 hours.
Safety
Safety evaluation was based on the incidence, seriousness, intensity and causal relationship of treatment-emergent adverse events (TEAEs). AEs were assessed throughout the study. Safety was evaluated by the assessment of clinically significant changes postdose versus the baseline in the physical examination, vital signs (VSs; blood pressure and heart rate) and laboratory safety tests (haematology, biochemistry and urinalysis). Any patient who prematurely withdrew after having received study medication was encouraged to undergo the end of study visit.
Statistical analysis
The primary efficacy variable was analysed on the per-protocol (PP) and intention-to-treat (ITT) populations to assess the non-inferiority hypothesis using analysis of covariance (ANCOVA) with one-sided significance level of 2.5% for testing the differences in treatment efficacy, as quantified by TOTPAR6, between TRAM/DKP and TRAM/paracetamol. The ANCOVA model included terms of treatment and the baseline PI (NRS) as covariates. In case of non-inferiority being confirmed, the superiority of TRAM/DKP versus TRAM/paracetamol was tested on the ITT population. Superiority of TRAM/DKP and TRAM/paracetamol versus placebo was evaluated in order to confirm the model sensitivity on the ITT population. Non-inferiority hypothesis was satisfied if the lower limit of the CI for the estimated difference between TRAM/DKP and TRAM/paracetamol was greater than a non-inferiority margin of 20% of the estimated mean of TRAM/paracetamol.
Secondary efficacy analysis SPID6 was analysed for non-inferiority with the possibility to switch for superiority analogously to the primary efficacy variable. All the other secondary efficacy variables were descriptively analysed and tested for the superiority of TRAM/DKP, when applicable, through ad hoc inferential analyses, as follows: NRS-PI, SPID (excluded SPID6), %max SPID, TOTPAR (excluded TOTPAR6), %max TOTPAR (continuous variables) were analysed by ANCOVA model as for the primary efficacy variable or a repeated measure model if more than one value per patient; VRS-PAR and PGE (categorical variables) were analysed by Wilcoxon rank-sum test; percentage of patients who required RM, and percentage of patients achieving at least 50% max TOTPAR or 30% of PI reduction, with confirmed FPPAR or MPAR were tested using a χ2test. Time to use RM, time to FPPAR, confirmed FPPAR and MPAR were assessed using a log-rank test. All analyses were performed in SAS V.9.3 (SAS).
Imputation
The method of last observation carried forward was applied among patients who missed more than one consecutive data input; otherwise the missing value was replaced by the mean of the two non-missing data collected, respectively, before and after the missing one. This procedure was applied to all efficacy outcomes. If PI was missed at t0, the value recorded during qualification procedure was used as a baseline. After RM intake, PI returned to its baseline (t0) level and PAR to zero (‘no relief’) for all subsequent time points (ie, baseline observation carried forward).
Safety analysis
AEs were coded using the MedDRA dictionary. The incidence of each TEAE was summarised by system organ class, preferred term and treatment. Clinically significant abnormal findings in VS and physical examination were listed by treatment. Safety variables were analysed by descriptive statistics and were run on the safety population.
Definition of analysis population
The ITT population consisted of all patients randomised; safety population of all patients who received study drug; PP population of all patients of the ITT who did not experience relevant protocol deviation related to efficacy endpoints of primary interest.
Patient involvement
No patients were involved in setting the research question or the outcome measures, nor were they involved in developing plans for recruitment, design or implementation of the study. No patients were asked to advise on interpretation or writing up of results. There are no specific plans to disseminate the results of the research to study participants or the relevant patient community beyond the usual channels of publication.
Role of the funding source
The study sponsor (Menarini Group) contributed to the study design, data analysis and manuscript preparation. MH confirms that he had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Of the 792 patients screened, 654 were randomised and received study treatment, one patient was excluded from any analysis being <18 years old (figure 1, table 1).
Patients by treatment group

Participant flow chart of study. ITT population consisted of all patients randomised; safety population of all patients who received study drug; PP population of all patients of the ITT who did not experience relevant protocol deviation related to efficacy endpoints of primary interest. *One patient excluded from analysis being aged less than 18 years. ITT, intention to treat; PP, per protocol; TRAM/DKP, tramadol/dexketoprofen.
The safety population comprised 653 patients as did the ITT group. Patients were enrolled at hospital clinics and dental surgeries across five European countries (66 patients in Hungary, 180 in Italy, 127 in Poland, 141 in Spain and 139 in the UK).
Efficacy analyses for non-inferiority were performed on the PP population, including a total of 620 randomised patients as 33 participants were excluded due to major protocol deviations related to efficacy endpoints of primary interest. Patients’ assignment to the different analysis populations occurred before the study blind was broken. One patient in the TRAM/paracetamol arm was lost at follow-up and did not attend end of study visit (figure 1). Demography and baseline characteristics of different treatment groups were comparable (table 2).
Patient demographic and baseline characteristics by treatment group (ITT population)
The mean (SD) age was 26.8 (7.48) years (range 18–52) in TRAM/DKP, 27.1 (8.13) years (range 18–63) in TRAM/Paracetamol; 26.5 (7.67) years (18–59) in placebo arm. Percentages of women were 58.5% in TRAM/DKP, 60.3% in TRAM/Paracetamol and 59.5% in placebo arm (table 2). Patients reported mean (SD) PI values at baseline of 5.7 (1.36); 5.5 (1.34) and 5.6 (1.30) in the TRAM/DKP, TRAM/paracetamol and placebo arm, respectively (table 2).
Efficacy results
Primary endpoint
Overall, the combination TRAM/DKP showed the greatest sustained analgesia during the 6-hour postdose period as demonstrated by the TOTPAR6 primary endpoint, compared with the TRAM/paracetamol and placebo groups. Since non-inferiority was confirmed, the ITT population was used to perform superiority analyses on the primary endpoint, as prespecified. The mean (SD) TOTPAR6 reported by patients in the TRAM/DKP group was 13 (7.0), while those in TRAM/paracetamol and placebo groups were 9.2 (7.7) and 1.9 (3.9), respectively, demonstrating that the combination TRAM/DKP was statistically superior (p<0.0001) to TRAM/paracetamol (table 3 and figure 2). The results of the analysis conducted on ITT population were consistent with those conducted on the PP, therefore, the model sensitivity was confirmed.
Summary of descriptive statistics TOTPAR, % max TOTPAR SPID and % max SPID over 2, 4, 6 and 8 hours

Mean TOTPAR at 6 hours (primary endpoint) and at 2, 4 and 8 hours for TRAM/DKP, TRAM/paracetamol and placebo with PAR was measured on a 5-point Verbal Rating Scale (0=‘no relief’ to 4=‘complete relief’). *Statistically significant comparison of TRAM/DKP versus TRAM/paracetamol (p<0.0001). PAR, pain relief; TRAM/DKP, tramadol/dexketoprofen; TOTPAR, total pain relief.
Secondary endpoints
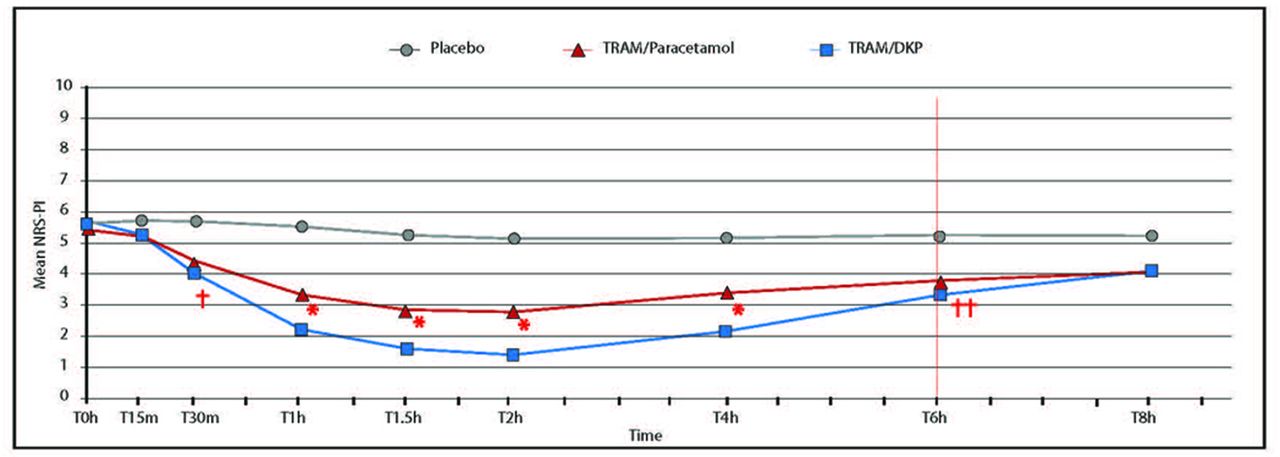
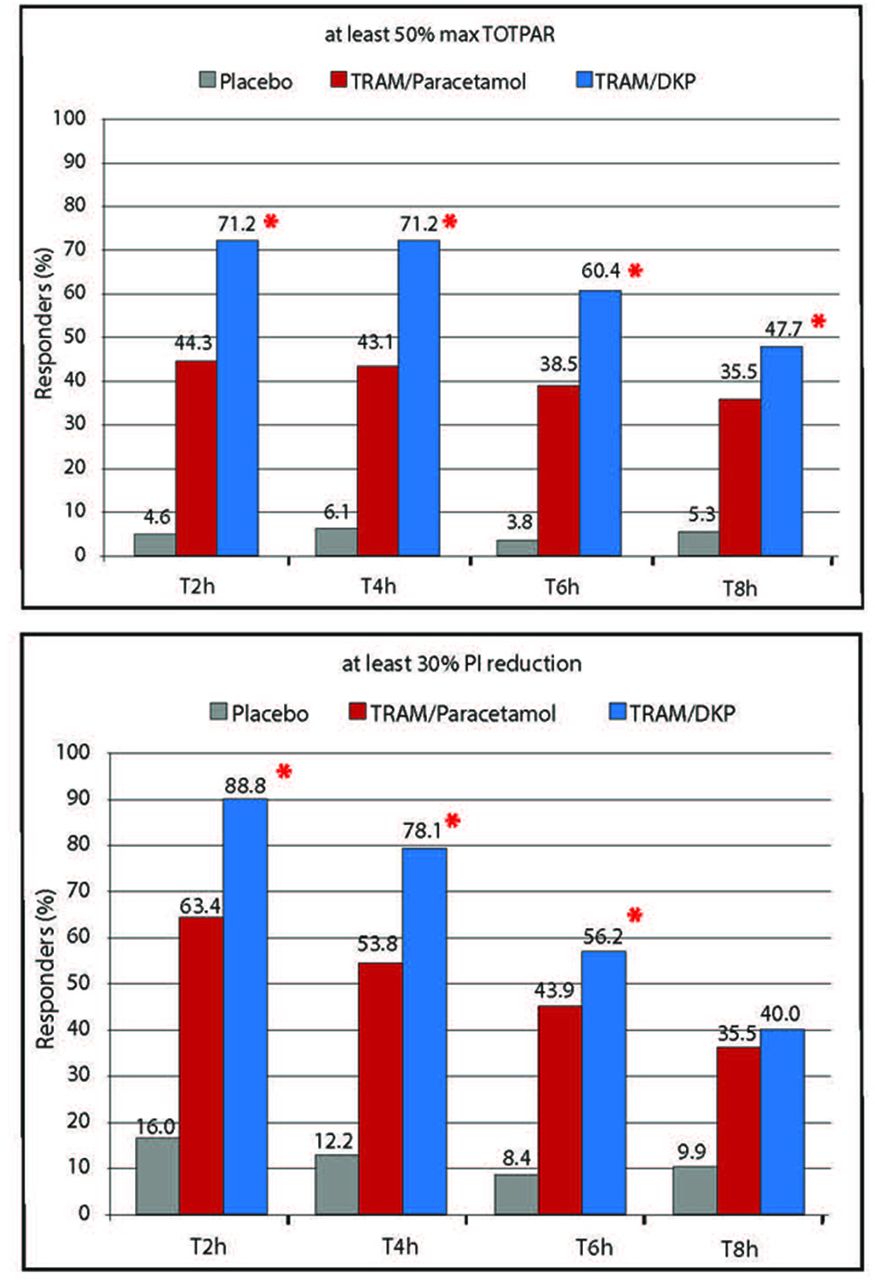
The time course of the mean PAR and PI scores showed that TRAM/DKP provided a more rapid onset of action compared with TRAM/paracetamol as differences were statistically significant already 30 min postdose. Furthermore, patients in the TRAM/DKP arm constantly had greater analgesia and lower PI, in a statistically and clinically significant fashion, at each prespecified time points until 6 hours after drug intake, in comparison to those in the comparator arm (figures 3 and 4). In addition, the PI analysis with repeated measures indicated superiority of the combination TRAM/DKP in comparison to TRAM/paracetamol during the overall 8-hour period of assessment (p=0.0009). The superiority of TRAM/DKP was also confirmed by mean values of the main secondary efficacy variable SPID6 (table 3). Analysis of summary efficacy measures including TOTPAR2, 4 and 8; SPID2, 4 and 8; % max TOTPAR over 2, 4, 6 and 8 hours; % max SPID over 2, 4, 6 and 8 hours provided further confirmation of the superiority of TRAM/DKP versus TRAM/paracetamol (p<0.0001) (table 3, figures 2 and 5). Regarding percentage of responders (patients achieving at least 50% of maxTOTPAR), the best results were detected in patients treated with TRAM/DKP, who were considered responders: 71.2%, 2 hours and at 4 hours, 60.4% at 6 hours and 47.7% at 8 hours. While responders in the TRAM/paracetamol group were 44.3%, 43.1%, 38.5% and 35.5% at 2, 4, 6 and 8 hours, respectively (p<0.01 for each time point) (table 4). In addition, when responders were defined as subjects who achieved at least 30% of PI reduction versus baseline, the best results were observed with TRAM/DKP versus TRAM/paracetamol at 2, 4 and 6 hours (p<0.01) (table 4, figure 6).

Time course of mean PAR over 8 hours for TRAM/DKP, TRAM/paracetamol and placebo with PAR measured on a 5-point Verbal Rating Scale (0=‘no relief’ to 4=‘complete relief’). The area under the curve for pain relief at a given time point corresponds to TOTPAR at the same time point. *Statistically significant TRAM/DKP versus TRAM/paracetamol (p<0.0001); †Statistically significant TRAM/DKP versus TRAM/paracetamol (p<0.0006); ††Statistically significant TRAM/DKP versus TRAM/paracetamol (p<0.00086). PAR, pain relief; TRAM/DKP, tramadol/dexketoprofen; TOTPAR, total pain relief.

Time course of mean PI over 8 hours for TRAM/DKP, TRAM/paracetamol and placebo with PI measured on 11-point Numerical Rating Scale (NRS) ranging from 0 (no pain) to 10 (worst pain). *Statistically significant TRAM/DKP versus TRAM/paracetamol (p<0.0001); †Statistically significant (p=0.0021); ††TRAM/DKP versus TRAM/paracetamol (p=0.0052). PI, pain intensity; TRAM/DKP, tramadol/dexketoprofen.
Percentage of responders to treatment over 2, 4, 6 and 8 hours (ITT population)

Percentage of max TOTPAR at 2, 4, 6 and 8 hours for TRAM/DKP, TRAM/paracetamol and placebo with PAR measured on a 5-point Verbal Rating Scale (0=‘no relief’ to 4=‘complete relief’). *Statistically significant TRAM/DKP versus TRAM/paracetamol (p<0.0001). PAR, pain relief; TRAM/DKP, tramadol/dexketoprofen; TOTPAR, total pain relief.

Percentage of responder patient by treatment and time points. Response defined as at least 50% max TOTPAR or 30% PI reduction. *Statistically significant TRAM/DKP versus TRAM/paracetamol (p<0.01). PI, pain intensity; TRAM/DKP, tramadol/dexketoprofen; TOTPAR, total pain relief.
The onset of analgesia, evaluated with the double stopwatch method, was significantly faster in the TRAM/DKP group in comparison to TRAM/paracetamol group considering all the relevant secondary endpoints (FPPAR, confirmed FPPAR and MPAR) measured within different time periods (30 min, 1 hour and 2 hours postdose). Significantly more patients in the TRAM/DKP group reported a confirmed FPPAR compared with patients in the TRAM/paracetamol group: 76.9% vs 60.3% at 30 min, 90.0% vs 72.5% at 1 hour, and 90.4% vs 72.9% at 2 hours (p<0.0001) (table 5, figure 7).
Proportion of patients achieving FPPAR, confirmed FPPAR and MPAR at prespecified time interval of 30 min, 1 hour and 2 hours using stopwatch (ITT population)

Percentage of patients achieving confirmed FPPAR within 30 min, 1 hour and 2 hours. TRAM/DKP versus TRAM/paracetamol. *P<0.0001. FPPAR, first perceptible pain relief; TRAM/DKP, tramadol/dexketoprofen.
The median (95% CI) times to confirmed FPPAR and MPAR after single dose of TRAM/DKP were 22 (18 to 24) and 41(33 to 45) min, respectively; while the ones in the TRAM/paracetamol group were 27 (23 to 27) and 57 (49 to 57) min, respectively. Log-rank test between TRAM/DKP and active comparator showed a statistically significant difference (p<0.0001) (figures 8 and 9). PGE data also showed a better performance of the combination containing DKP and TRAM over the comparator. Significantly more patients in TRAM/DKP arm (80.8%) rated the study medication as ‘good’, ‘very good’ or ‘excellent’, than that in the TRAM/paracetamol (56.6%) and placebo (15.3%) groups (table 6, figures 10 and 11).
Summary of Patient Global Evaluation scores measured on a 5-point VRS (1=poor to 5=excellent) at the end of assessment period (ITT population)

Kaplan-Meier estimation of time to patient report of confirmed first perceptible pain relief (FPPAR) represents the cumulative percentage of patients reporting confirmed FPPAR over the time (in minutes) in the TRAM/DKP arm and TRAM/paracetamol arm. TRAM/DKP, tramadol/dexketoprofen.
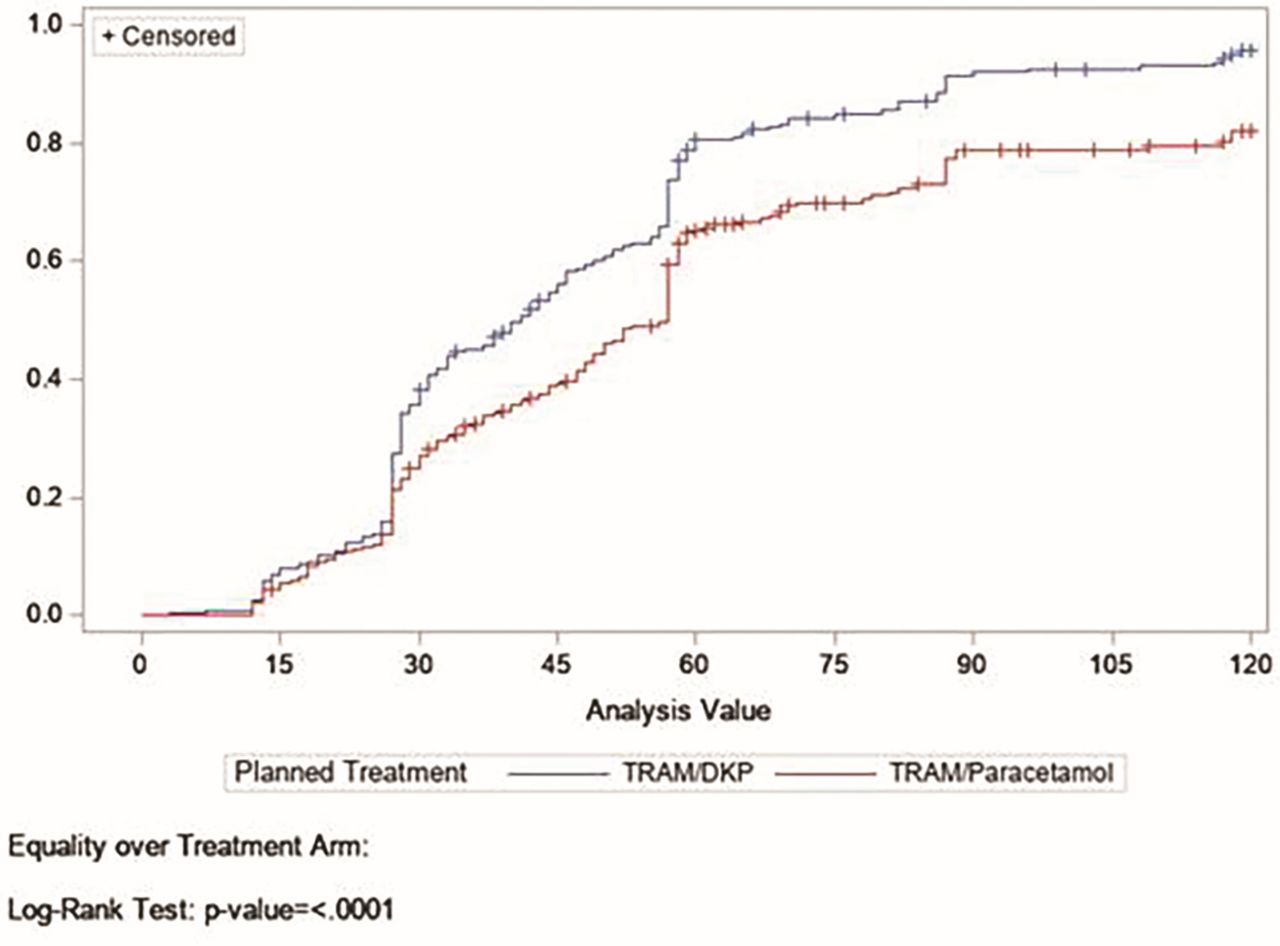
Kaplan-Meier estimation of time to patient report of meaningful pain relief (MPAR) represents the cumulative percentage of patients reporting MPAR over the time (in minutes) in the TRAM/DKP arm and TRAM/paracetamol arm. TRAM/DKP, tramadol/dexketoprofen.

Mean scores of patients’ global evaluation by treatment arm assessed on 5-point Verbal Rating Scale (1=poor to 5=excellent). *Statistically significant TRAM/DKP versus TRAM/paracetamol (p<0.0001). TRAM/DKP, tramadol/dexketoprofen.

Percentage of subjects who rated the treatment as poor, fair, good, very good or excellent on patients’ global evaluation by treatment arm. TRAM/DKP, tramadol/dexketoprofen.
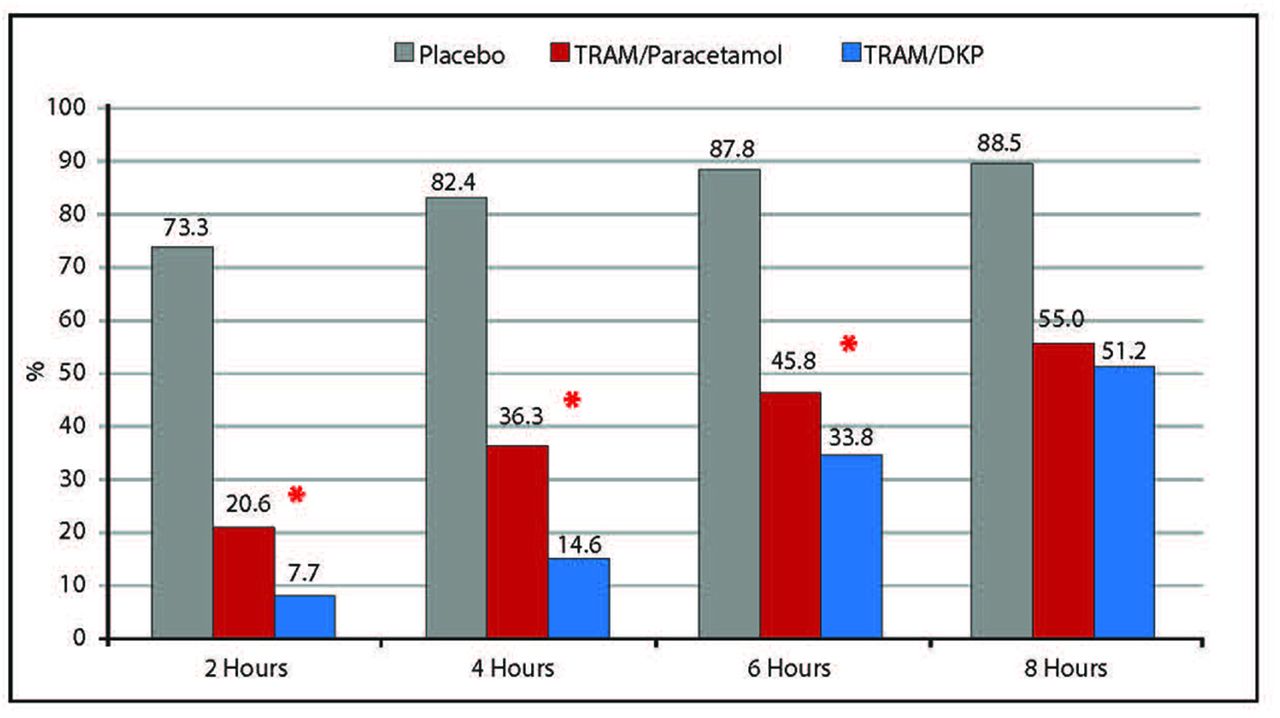
RM was required by significantly fewer patients in the TRAM/DKP group (7.7% within 2 hours; 14.6% within 4 hours; 33.8% within 6 hours) than in the TRAM/paracetamol group (20.6% within 2 hours; 36.3% within 4 hours; 45.8% within 6 hours) (p<0.01). The majority of patients randomised to placebo took RM (73.3% within 2 hours; 82.4% within 4 hours; 87.8% within 6 hours). Patients in TRAM/paracetamol group used RM more quickly than patients in TRAM/DKP group (p=0.0373) (figures 12 and 13).

Percentage of patient taking first rescue medication within 2, 4, 6 and 8 hours after drug intake. * Statistically significant TRAM/DKP versus TRAM/paracetamol (p<0.01). TRAM/DKP, tramadol/dexketoprofen.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier estimation of time to RM intake represents the cumulative percentage of patients using RM over the time (in minutes) in the TRAM/DKP arm and TRAM/paracetamol arm. RM, rescue medication; TRAM/DKP, tramadol/dexketoprofen.
Safety
Overall, 53 patients (8.1%) experienced one or more adverse drug reactions (97 ADRs in total) but none of the ADRs were considered to be serious (table 7).
Overview of ADRs, by treatment and overall
No clinically relevant differences were identified in ADRs incidences between treatment groups. Overall, the most common ADRs were: vomiting (3.8%), nausea (3.4%), dizziness (2.9%) and somnolence (2.1%) (table 8). No deaths or other significant ADRs occurred. There were no clinically relevant changes in the VSs or physical examination versus baseline. Overall, TRAM/DKP was safe and well tolerated, presenting a safety and tolerability profile fully in line with that observed in previous clinical experience.
Overview of adverse drug reactions by MedDRA system organ class (SOC) and preferred term (PT)
Discussion
This study shows that TRAM/DKP provides an effective analgesia for the control of moderate to severe pain following removal of impacted lower third molar and is superior to TRAM/paracetamol in terms of rapidity of onset and intensity of analgesia. Statistically and clinically significant superiority of TRAM/DKP over TRAM/paracetamol was demonstrated with an overall consistency not only at the primary endpoint(ie, TOTPAR6), but also at all different outcome measures adopted in the study. Study participants receiving TRAM/DKP combination reported lower pain scores already at the 30 min time point after drug administration.
Limitations
The main limitation is that this was a single-dose study and as such assessed only the short-term analgesic efficacy of TRAM/DKP in comparison with TRAM/paracetamol. In addition, although it has been proposed that systematic differences in the estimate of analgesic efficacy between dental and postsurgical pain models are unlikely,26 and efficacy in the dental model is highly predictive of efficacy in later stage models in drug development,12 further studies with similar design applied in other acute pain conditions are warranted.
Generalisability
The results of this head-to-head trial can guide physicians in choosing the adequate analgesic medication to optimise clinical outcomes in the treatment of a wide range of acute pain. This is particularly true in a moment in which there is a clear tendency to abandon the early treatment with strong opioids, in patients suffering from acute moderate to severe pain. Results of the present trial reinforce the clinical benefit of TRAM/DKP that emerged in the whole developmental clinical programme of this fixed combination that involved some 1900 patients with moderate to severe acute pain assessed using well-established human models of acute visceral and somatic moderate to severe pain.11–16
Interpretation
Fast action is an important feature for an analgesic intended to be used in the treatment of acute pain.29 The earlier onset of analgesic efficacy with TRAM/DKP compared with TRAM/paracetamol may be attributable to the rapid and high bioavailability of DKP,29 30 that favours the rapid onset of action of the combination. Better analgesic efficacy was maintained consistently over the entire observation period, confirming the sustained analgesic action observed in previous clinical trials assessing efficacy following third molar extraction, abdominal hysterectomy and total hip arthroplasty.11 13 14 The greater analgesic efficacy of TRAM/DKP results from the balanced and synergistic combination of peripheral and central analgesia, complemented with an anti-inflammatory action.13 The combination TRAM/paracetamol is substantially lacking in anti-inflammatory activity, as paracetamol only inhibits the synthesis of prostaglandins in the central nervous system and peripherally blocks pain impulse generation, but unlike NSAIDs, is not a peripheral COX inhibitor.31 32 It can be hypothesised that this difference could have contributed to the observed reduced analgesic efficacy.
The two combinations showed clinically comparable safety profiles. Gastrointestinal disorders including abdominal discomfort, diarrhoea, nausea and vomiting were reported with similar frequencies between the two groups of patients who received active intervention. Adverse reactions regarding the nervous system disorders were also observed in a similar proportion in both arms, although a trend towards improved tolerability could be detected in patients receiving TRAM/DKP, as compared with patients treated with TRAM/paracetamol.
Conclusion
This study has confirmed that TRAM hydrochloride/DKP trometamol 75/25 mg oral fixed combination is effective and superior over TRAM hydrochloride/paracetamol 75/650 mg in relieving moderate to severe acute pain following removal of impacted lower third molar. TRAM hydrochloride/DKP trometamol 75/25 mg oral fixed combination shows faster onset of effect, greater and durable analgesia, together with a favourable safety profile. The rapid onset of analgesic effect of DKP, with its anti-inflammatory activity, associated to the sustained action of TRAM, makes this combination a valuable tool to achieve multimodal analgesia.
References
Footnotes
Contributors Study design: CG-E, MH, GV and AM. Patient recruitment and data collection: CG-E, TD, SM, EG and TBZ. Data analysis: MH, GV, AM and CG-E. Preparation of the manuscript: CG-E, MH, GV and AM. All authors contributed to the development of interim and final drafts, and read and approved the final manuscript.
Funding This study was funded by Menarini Group.
Competing interests MH reports personal fees as a consultant for educational symposia by Menarini Group. CG-E reports personal fees from Menarini Group, outside the submitted work. TD reports grants from Menarini Group, during the conduct of the study; grants and personal fees from Institut Biochimique SA, outside the submitted work. AM, SM, EG, TBZ and GV declare no conflict of interest. We attest that we have obtained appropriate permissions and paid any required fees for use of copyright protected materials.
Ethics approval The study approved by all the concerned Competent Authorities and Ethics Committees, (Sponsor Code DEX-TRA-06).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The relevant anonymised patient level data are available on reasonable request from the authors.
Correction notice Since this article was first published online changes have been made. A separate correction notice will be published listing these changes.
Patient consent for publication Not required.