Article Text

Abstract
Objectives To determine the feasibility of undertaking a randomised controlled effectiveness trial evaluating the use of a perineural catheter (PNC) after major lower limb amputation with postoperative pain as the primary outcome.
Design Randomised controlled feasibility trial.
Setting Two vascular Centres in South Wales, UK.
Participants 50 patients scheduled for major lower limb amputation (below or above knee) for complications of peripheral vascular disease.
Interventions The treatment arm received a PNC placed adjacent to the sciatic or tibial nerve at the time of surgery, with continuous infusion of levobupivacaine hydrochloride 0.125% for up to 5 days. The control arm received neither local anaesthetic nor PNC. Both arms received usual perioperative anaesthesia and postoperative analgesia.
Primary and secondary outcome measures The primary outcomes were the proportion of eligible patients who were randomised and the proportion of recruited patients who provided primary effectiveness outcome data. Secondary outcomes were: the proportion of recruited patients reaching 2 and 6 month follow-up and supplying pain data; identification of key cost drivers; development of an economic analysis framework for a future effectiveness trial; identification of barriers to recruitment and site set-up; and identification of the best way to measure postoperative pain.
Results Seventy-six of 103 screened patients were deemed eligible over a 10 month period. Fifty (64.5%) of these patients were randomised, with one excluded in the perioperative period. Forty-five (91.3%) of 49 recruited patients provided enough pain scores on a 4-point verbal rating scale to allow primary effectiveness outcome evaluation. Attrition rates were high; 18 patients supplied data at 6 month follow-up. Costs were dominated by length of hospital stay. Patients and healthcare professionals reported that trial processes were acceptable.
Conclusions Recruitment of patients into a trial comparing PNC use to usual care after major lower limb amputation with postoperative pain measured on a 4-point verbal rating scale is feasible. Evaluation of longer-term symptoms is difficult.
Trial registration number ISRCTN: 85 710 690. EudraCT: 2016-003544-37.
- amputation
- perineural
- pain
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
PLACEMENT comprises a comprehensive feasibility study, where all aspects of an effectiveness trial have been undertaken including multicentre recruitment, randomisation, data collection and follow-up.
In addition, we have performed qualitative studies to assess barriers to recruitment, developed a framework for a pragmatic economic evaluation and identified a suitable strategy for measuring postoperative pain.
Limitations of the study are that it lacks the power to assess the effectiveness of the intervention based on this feasibility study alone, and it does not assess the use of a placebo arm.
Background
Despite advances in peripheral revascularisation, 1% to 2% of patients with peripheral arterial disease (PAD) undergo major lower limb amputation.1 2 Postoperatively, many amputees experience acute pain, and in the long-term chronic stump pain (CSP) and phantom limb pain (PLP) can affect as many as 80% of patients.3 4 Treating both acute and chronic pain is difficult,5 and there is some evidence that suboptimal control of perioperative pain may increase rates of PLP or CSP.4 6 The UK National Confidential Enquiry into Patient Outcome and Deaths review of lower limb amputations found that epidural or intravenous patient controlled opioid analgesia were the most commonly employed methods of managing analgesia postoperatively, but that only 37% of patients experienced good acute pain control.7 While opioids may provide good analgesia, they have associated side effects, including nausea, vomiting,8 constipation,9 sedation and pruritus.10 Their use has also been shown to increase both hospital costs and length of stay.11 12 Lumbar spinal epidural anaesthesia is likely to be the optimal strategy for managing postoperative pain in this cohort.13–15 However, due to use of anticoagulants or antiplatelet agents, or the presence of an active infection, epidural anaesthesia is contraindicated over 60% of the time.15
The use of perioperative and postoperative local anaesthetic infiltration and infusion to the surgical site is recognised as a useful adjunct in postoperative pain relief.16 Peripheral wound catheters (PWCs) are thin catheters placed at the time of surgery in the surgical field, of which perineural catheters (PNCs) are a specific type, placed adjacent to major nerves. A continuous infusion of local anaesthetic can thus be delivered directly to the perineural space.17 PWC usage has been associated with improved pain scores, reduced opioid use, reduced length of hospital stay and increased patient satisfaction,18 with few adverse events.19 20
PNCs have been used for major amputations, inserted adjacent to either the sciatic or tibial nerve (for above knee amputations (AKAs) or below knee amputations (BKAs), respectively), since at least 1991.21 A systematic review of PNC use in lower limb amputation showed patients receiving a PNC required less opioid analgesia compared with controls, although no significant difference was noted in pain scores.22 However, the quality of included papers was low, studies were small and comparison groups were non-standardised. Other papers excluded from the meta-analysis or published in the interim have generally suggested that PNCs reduce postoperative pain.23–27 During trial set-up we performed a survey of vascular Centres in the UK which demonstrated that almost exactly half of surveyed centres were using PNCs ‘most of the time’ or ‘all of the time’, suggesting there was equipoise within the vascular community as a whole regarding their value in amputees.
While there are limited data to suggest PNCs are beneficial in reducing postoperative pain and opioid use, the overall quality of evidence is very low, making a contemporaneous randomised controlled trial (RCT) necessary to establish its value in pain relief, opioid use, quality of life (QoL)28 and overall cost. Given the very low-quality evidence available, a feasibility study was deemed a necessary first step in order to inform trial design and sample size calculation. The PLACEMENT trial was thus designed as a parallel group, randomised controlled feasibility study to assess the feasibility of conducting an effectiveness trial evaluating PNC insertion with a continuous local anaesthetic infusion, inserted at the time of amputation, on short and medium term postoperative outcomes.29
Materials and methods
Trial design
PLACEMENT was a pragmatic randomised unblinded 1:1 feasibility study, undertaken at the Royal Gwent Hospital, Aneurin Bevan University Health Board and Morriston Hospital, Abertawe Bro Morgannwg University Health Board, Wales. The protocol is available as a online supplementary file. The study is reported in line with Consolidated Standards of Reporting Trials guidelines.30 Fifty adult patients undergoing major lower limb amputation, either BKA or AKA were randomised to receive the intervention, described below, or usual care.
Supplemental material
Inclusion and exclusion criteria
Patients were suitable for inclusion if they were aged 18 years or older, undergoing elective or emergency BKA or AKA for complications of PAD, were able to assess pain using a Verbal Rating Scale (VRS), and (for women of childbearing potential) were willing to undergo a pregnancy test before the trial and agree to either use a highly effective method of contraception or abstain from sexual intercourse until at least 7 days after their amputation. Patients with a life expectancy of greater than 2 weeks were suitable, which was done to minimise early patient attrition in those deemed very high risk for surgery.
Patients undergoing digital, metatarsal, tarsal or through knee amputation, disarticulation of the hip, hindquarter amputation or simultaneous bilateral amputations were excluded. Patients unable to provide consent due to incapacity (as defined by the Mental Capacity Act 2005), and vulnerable or protected adults, were excluded. Patients with an allergy or intolerance to any of the substances in the PNC, or local anaesthetic agents, or chronically taking class IB anti-arrhythmic agents or local anaesthetic agents, were excluded, as were pregnant females and patients expected to be managed in the intensive care unit (ICU) postoperatively and be sedated for more than 24 hours. Patients undergoing more than one major amputation were not permitted to be enrolled twice.
In addition, patients who were initially eligible for PLACEMENT could become ineligible if the major amputation was not performed, the appropriate nerve was not identified or if due to perioperative or intraoperative instability, the patient required admission to the ICU and was likely to be sedated for more than 24 hours, or not expected to survive more than 2 weeks.
Trial procedures
Intervention arm
Participants randomised to the treatment arm received a PNC placed at the time of the amputation (either to the sciatic or tibial nerve) with a continuous infusion of levobupivacaine hydrochloride 0.125% (2.5 to 15 mg/hour) for up to 5 days, in addition to usual care (described below). All surgeons were assessed and trained prior to trial enrolment to ensure PNC placement was standardised. During the amputation the nerve was identified. An epidural catheter tube was used as the PNC and placed via a Tuohy needle in the perineural space to ensure that the tip lay 10 to 20 cm cranially from the amputated end. The catheter end was removed through the skin distant to the wound edge and secured. Division of the nerve was then performed under tension.
Local anaesthetic infusion was delivered via an elastomeric pump (AutoFuser, ACE Medical, Korea), and started in the immediate postoperative period, with standard monitoring of the pump by trained ward nursing staff and members of the acute pain team, as per local hospital policy.
Control arm
Participants randomised to the control arm received usual care. This consisted of standard anaesthetic and postoperative analgesia, with daily review by a specialist pain team. Preoperative epidural analgesia, spinal analgesia, regional nerve blocks or PWCs were permitted in either arm. Patient-controlled analgesia was used when appropriate after discussion with patients. Patients were cared for by ward staff familiar with patient-controlled analgesia and use of the elastomeric pump.
Qualitative assessment of trial processes and patient experience
In order to evaluate trial processes and acceptability of the intervention, semi-structured interviews were undertaken to explore the experiences of trial participants and health professionals. Interviews were conducted by experienced qualitative health researchers (SM, MM, HS) who were not involved in the delivery of the trial.
Twenty semi-structured interviews were conducted with 14 trial participants (seven in the intervention arm, seven in the control arm) purposefully sampled to include participants at both study sites, from both arms of the trial, while also taking into consideration variation in gender, age and amputation type (below and above knee). Interviews were carried out at two time points; interview 1 within the postoperative period and up to 1 month following amputation (n=10); interview 2 between 6 and 16 months following amputation (n=10). Six participants completed an interview at both time points (forming paired interviews). Interviews with trial participants took place between May 2017 to July 2018 and explored patients’ experiences and perceptions of postoperative pain management, together with understanding and acceptability of trial processes. Interviews were also conducted with 10 health professionals directly involved in the trial and purposefully sampled to include variation in role (eg, vascular surgeons, research nurses, etc) and representation across both study sites. Interviews took place between February 2018 to April 2018 and focused on the delivery of the intervention in the real healthcare setting, exploring fidelity, feasibility and acceptability. Topic guides were developed by the qualitative researchers, discussed within the Trial Management Group (TMG) and reviewed again following early interviews. All interviews were audio recorded with the permission of the interviewee, transcribed verbatim, anonymised and uploaded to NVivo 11 for analysis. A systematic five-stage framework approach was used in order to facilitate exploration and comparison of the data across cases and themes. The first three stages ‘familiarisation with data’, ‘identifying a thematic framework’ and ‘indexing’, are common to other forms of qualitative analysis. The fourth stage, ‘charting’, involves retrieving the coded data and producing summaries of interviewees’ talk on each theme and visually arranging it in a table to build an overall picture of the whole data set. The fifth stage, ‘mapping’, involves using the charts to map and interpret the data set as a whole and connect with the original research objectives. SM developed a thematic framework using a deductive approach to incorporate the research objectives and topic guide, and an inductive approach to ensure that new themes, which had not been pre-empted and initiated from the participants themselves, were identified in the data. The thematic framework was developed and revised after regular discussion with LB-H. Transcripts were double-coded until consensus was reached through regular discussion to enhance analytic reliability and validity.
Outcomes
Feasibility outcomes
As a feasibility study, the primary outcomes were designed to assess if running an effectiveness trial investigating the effect of PNC placement with local anaesthetic infusion on immediate postoperative pain is possible and appropriate, and to establish the best way to undertake such a trial. The primary outcomes were therefore concerned with patient recruitment and outcome assessment.
The primary recruitment outcome was the proportion of eligible patients who were recruited.
The primary assessment outcome was the proportion of recruited patients who provided adequate putative primary effectiveness outcome data, using the various methods of postoperative pain assessment (see below).
Secondary outcomes included:
Recruitment and retention – the proportion of eligible patients who provided informed consent; the proportion of those recruited who reached 2 and 6 month follow-up; the overall time to complete recruitment.
Assessment of pain – identify appropriate methods and tools for collecting postoperative pain data.
Sample size calculation – sample size estimation for a future effectiveness trial, based on effect size estimates.
Secondary outcome assessment – identify what secondary outcome measures (both short-term and long-term measures) are important, and how best to assess them.
Economic evaluation – identify key cost drivers and develop a robust framework for economic evaluation in a future effectiveness trial.
Trial management and quality control – evaluation of trial processes, from site set-up to final data collection, as detailed in the ‘Qualitative assessment of trial processes and patient experience’ section above.
Patient experience – develop a greater understanding of patient experience of pain and pain management, and the experience of being enrolled in the trial, as detailed in the ‘Qualitative assessment of trial processes and patient experience’ section above.
Effectiveness outcomes
Effectiveness outcomes were defined to provide a template for a future effectiveness trial.
Primary effectiveness outcome
One of the secondary feasibility outcomes of the study was to identify appropriate methods and tools to assess postoperative pain. We therefore defined two alternative primary effectiveness outcomes, according to which tool was found to be the most appropriate.
First, we collected pain scores on an 11-point (0 to 10) VRS, whenever suitably trained personnel were available, up to three times a day, with at least 4 hours between pain measurements, for the first five postoperative days.31 Second, we transformed any recorded 11-point VRS to a 4-point VRS (0 to 3; none/mild/moderate/severe) using a published algorithm32 and supplemented these with any 4-point VRS recorded on the routine nursing observations chart (the standard way of measuring pain in both trial centres) at times where there was no 11-point pain score available. The maximum potential pain recordings available were 17 (two from day of surgery, and three from the following 5 days).
The principle primary outcome was defined as the median 11-point VRS if at least nine pain scores were recorded, and ‘missing’ otherwise. This decision, taken by the TMG, and subsequently ratified ty the Trial Steering Committee (TSC), was to include all patient data records if more than 50% of possible pain score data points were reported. The alternative primary outcome was defined as the modal 4-point VRS if at least nine pain scores were recorded, and ‘missing’ otherwise.
Secondary effectiveness outcomes
The following secondary effectiveness outcomes were captured:
Overall Benefit of Analgesia Score (OBAS)33 preoperatively and once daily postoperatively for 5 days.
Self-completed Leeds Assessment of Neuropathic Symptoms and Signs scores (S-LANSS)34 preoperatively and on postoperative day 5.
Analgesia ‘Treatment failure’, defined as an 11-point VRS ≥4, or 4-point VRS of ‘Moderate’ or ‘Severe’ during the first 5 postoperative days
Opioid use measured preoperatively and once daily postoperatively for 5 days, converted to morphine equivalents.
Incidence of nausea or vomiting during the total inpatient stay.
Pain assessed by S-LANSS at 2 and 6 month follow-up.
PLP and CSP as assessed by the Groningen questionnaire35 at 2 and 6 month follow-up.
Health-related QoL assessed using EQ-5D-5L and Hospital Anxiety and Depression Scale (HADS; reported as both anxiety and depression scores) preoperatively, at 2 and 6 month follow-up.
Surgical site infection (SSI) rates classified as per the 2008 CDC/NHSN (Centers for Disease Control and Prevention/National Healthcare Safety Network) document.
Rate of successful identification of the nerve and PNC placement, as determined by the operating surgeon at the time of amputation.
Resource usage during the first six postoperative months.
Randomisation
Patients were randomised preoperatively or (preferably) intraoperatively. If preoperative randomisation was performed, the allocation was concealed from the surgeon and anaesthetist until the nerve was identified. Preoperative randomisation was only undertaken if the (unscrubbed) theatre staff were unable to perform the randomisation procedure. In this instance a single member of the unscrubbed theatre team completed the randomisation and recorded the outcome. Once the nerve was identified, the randomisation was given to the team. Randomisation was performed via a web-based remote randomisation programme, with telephone backup during working hours. Randomisation was minimised according to level of amputation (AKA vs BKA) and sex, and stratified by site.
Blinding
No individuals included in the study, including patients, surgeons, anaesthetists, nursing staff, outcome assessors or other healthcare professionals, were blinded in the study.
Sample size
The trial was designed to assess feasibility, rather than effectiveness, and a convenience sample of 50 patients was determined appropriate. Both primary feasibility outcomes were proportions, and this sample size allows for a total proportion to be conservatively estimated at 50% (ie, 25 out of 50) to within plus or minus 13.9% using a 95% CI.
Follow-up
Patients were contacted at 2 months (+/-2 weeks) and 6 months (+/-1 month) for follow-up, preferably via telephone. An experienced research nurse checked the hospital database to ensure the patient was not deceased, or an inpatient, prior to attempting contact. If the patient was unavailable or the phone call was unanswered, two further attempts were made to contact the patient after 2 to 7 days. Non-responders had questionnaires posted to them with a stamped addressed envelope for return.
Data captured at 2 and 6 months follow-up included data on PLP, CSP, neuropathic pain (S-LANSS), anxiety and depression (HADS), health-related quality of life (EQ-5D) and patient-reported healthcare resource usage. The Groningen questionnaire was used to capture data on CSP and PLP. The Groningen questionnaire provides continuous data on symptom burden, but with no standardised methods of dichotomising its presence or absence. A consensus was reached by the Trial Management Group to dichotomise patients into having CSP or PLP or not, depending on the results of the following questions. For CSP, we reviewed answers to the question: ‘In the last 6 weeks/months since your amputation, how often did your stump hurt badly enough to keep you from doing things you wanted to do?’. For PLP, we reviewedanswers to the question: ‘In the last 6 weeks/months since your amputation, how often did your phantom hurt badly enough to keep you from doing things you wanted to do?’. A result of ‘every day’ or ‘once per week’ was considered as CSP or PLP, respectively, and a result of ‘every two weeks’, ‘once per month’, ‘less than once per month’ and ‘never’ was considered as no CSP or PLP, respectively.
Management of adverse events
Adverse events were defined and reported according to the Medicines for Human Use (Clinical Trials) Regulations 2004 (SI2004/1031) and the subsequent amendment regulations (SI2006/1938) and reported and actioned as per Good Clinical Practice (GCP). These included infection around the PNC exit site and toxicity reactions.
Adverse events that are recognised complications of major lower limb amputation were also collected, such as infection, non-healing, debridement or surgical revision of the stump wound, myocardial infarction, acute coronary syndrome or arrhythmia not felt to be associated with toxicity to the Investigational Medicinal Product (IMP), pneumonia, pneumothorax, requirement for ventilation, tracheostomy, urinary retention, urinary tract infection, deep vein thrombosis, pulmonary embolism, stroke or transient ischaemic attack. These were considered as expected events which were causally unrelated to the IMP and trial intervention.
Data analysis
Analysis was carried out using SPSS 23.0 (descriptive statistics and non-parametric tests) and StataIC 13 (linear and logistic regression). Analysis was by intention-to-treat, with a planned secondary analysis of per-protocol treatment. All logistic and linear regression analyses were undertaken with the usual care arm as the reference and contained the following covariates: amputation type (above or below knee), sex (male or female), site (Royal Gwent Hospital or Morriston Hospital), patient age, opioid use at baseline, history of diabetes mellitus (yes or no), method of anaesthesia (with or without a continuous neuraxial anaesthetic infusion). The covariates included were agreed in the Statistical Analysis Plan by all members of the TMG and the TSC (who additionally held the role of an Independent Data Monitoring Committee). Where continuous outcomes could not be analysed using linear regression (even when transformed), a non-parametric Mann-Whitney U test was used to calculate a p value to assess the difference in medians between the two arms.
Sample size estimates
Sample size estimates for ordinal logistic regression ORs to achieve 90% power at the 5% significance level were estimated using the tool located at http://www.pmean.com/04/OrdinalLogistic.html.36 No imputation of missing data was performed.
Ethical and governance approval
The Trial Protocol was reviewed and approved by Wales Research Ethics Committee (REC) 3, recognised by the United Kingdom Ethics Committee Authority. Both hospital sites received Research and Development approval from the respective National Health Service Health Boards in Wales. A notice of no objection to the clinical trial notification was obtained from the Medicines and Healthcare products Regulatory Agency, and the trial was classified as a ‘type A’ Clinical Trial of Investigational Medical product, as both arms use procedures and medications which are already used in routine clinical practice.
Patient and public involvement
Patients and a lay representative have been integral in the design of PLACEMENT. Two lay representatives, one of whom is an amputee, have attended trial development and management meetings from inception and gave constructive input on the entire trial process. PLACEMENT was preceded by a public engagement group of amputees, led by an experienced qualitative researcher, to explore what factors were significant to amputees in the postoperative period, different experiences of postoperative pain relief (both with and without PNCs), and to explore if a trial examining PNC usage in the postoperative period would be acceptable to patients.
Results
Screening, recruitment, retention and long-term follow up
A total of 50 patients were randomised between February 2017 and November 2017 (figure 1). One hundred and three patients were screened for suitability, of whom 76 (73.8%) were deemed eligible. The most frequent reasons for being ineligible were the inability to assess pain using a VRS (n=13) or the inability to provide informed consent due to incapacity (n=6). Of the 76 eligible patients, 54 (71.1%) provided written informed consent. The most frequent reasons for patients not providing consent were because the patient declined (n=8), a trial surgeon was unavailable (n=7) or there was inadequate time to consent (n=4). Four patients consented were not randomised due to a change in surgery type (n=3) or the trial surgeon being unavailable (n=1). One patient, randomised preoperatively, developed severe respiratory compromise during anaesthetic induction, and was subsequently deemed ineligible and therefore excluded from the trial. Forty-nine patients were therefore available for analysis.
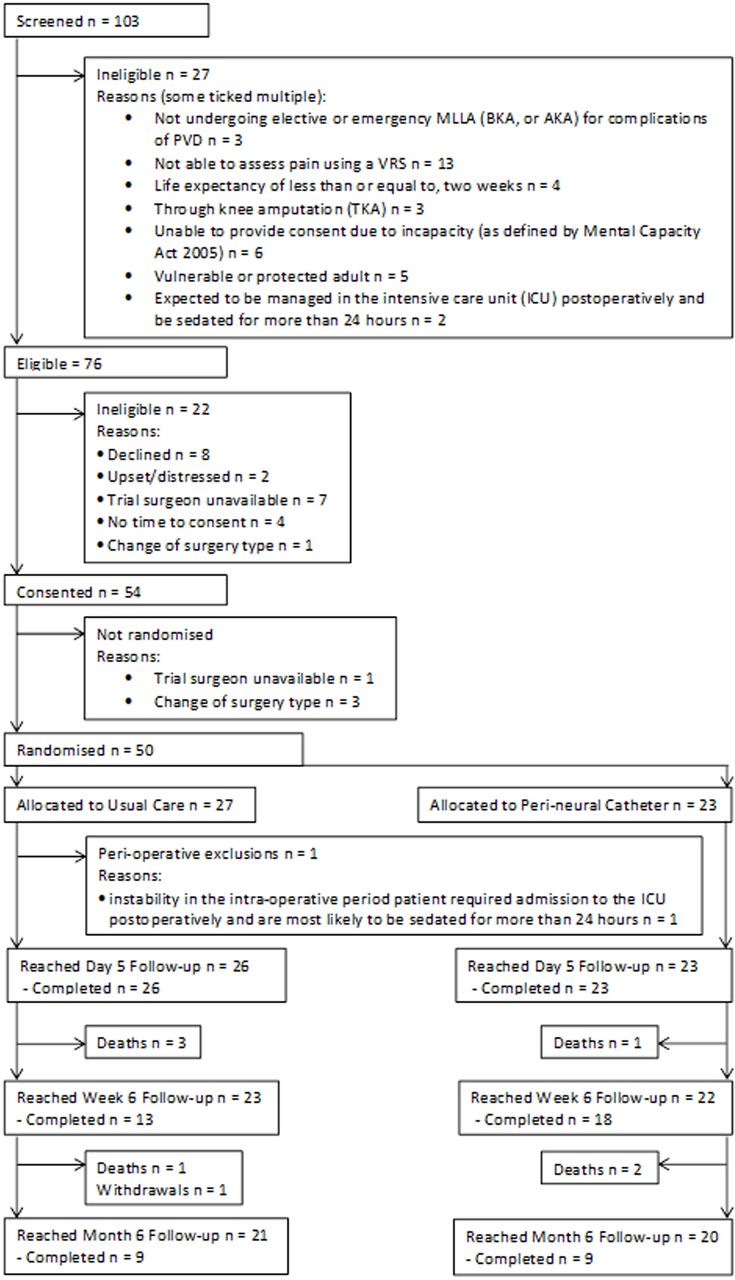
{kind=link}
Consolidated Standards of Reporting Trials patient flow diagram. AKA, above knee amputation; BKA, below knee amputation; PVD, peripheral vasculardisease; MLLA, major lower limb amputation; VRS, Verbal Rating Scale.
Twenty-three of the 49 patients (46.9%) received the intervention. One patient randomised to usual care received a PNC, but the remaining 48 patients received the correct treatment allocation. Baseline demographics are presented in online supplementary table 1, and baseline pain and opioid consumption data are presented in online supplementary table 2. No clinically relevant differences were noted in baseline pain or opioid usage between arms. The majority of patients were male (80%) with a mean age of 69.8 years. The BKA:AKA ratio was 1.63. Baseline data completion rates on pain, opioid use, anxiety and depression and costs are given in table 1.
Supplemental material
Percentage of completion of pain assessment tools and opioid consumption preoperatively (baseline) and in the immediate postoperative period
All patients completed 5 day follow-up. Although 45 patients survived to 2 months, only 31 provided outcome data. At 6 months, 41 (82%) were alive and had not withdrawn, but only 18/41 (39%) provided any outcome data.
Availability of primary effectiveness outcome data
There was significant missing data for the principle primary outcome, with 34 of 49 (69.4%) patients providing at least nine 11-point VRS pain scores. When these were transformed to the 4-point VRS and supplemented by routinely captured 4-point VRS, 45 of 49 (91.8%) patients provided at least nine pain scores and thus provided an alternative primary outcome.
Qualitative trial process interviews with healthcare professionals highlighted the difficulty in obtaining postoperative pain scores, particularly out of hours, when there were no research nurses, and where nurse-captured 4-point VRS scores were also sometimes missing. It was suggested that a reduction in the amount of data collected postoperatively would increase feasibility. Furthermore, both patients and healthcare professionals reported that using more than one pain scale was confusing for patients, with some giving conflicting scores across the two scales.
Availability of secondary outcome data
The completeness of data collection for other postoperative variables is given in table 1 and the percentage of data completion at follow-up is given in table 2. Missing data at 2 and 6 month follow-up were high. This was despite making every effort to contact patients in person (if they were an inpatient locally at the time), via telephone and by postal questionnaires. The time frame for data capture was deliberately broad to try and improve responses. Response rates to postal questionnaires, when sent, was low (9 of 32). Nurses were not sent to patients’ home address to increase data capture as it was felt that this would be unsustainable in any future effectiveness trial.
Percentage of completion rates at baseline, 2 and 6 months follow-up periods. No baseline Groningen data was captured
Healthcare professionals identified in qualitative process evaluation interviews that capturing data at 6 months was problematic as patients often could not be located; however, it was acknowledged that long-term follow-up is important to examine levels of chronic pain. It was highlighted that many of the follow-up questions could not be answered if patients were still in hospital. The time required to answer all questions was significant, and it was suggested that response rates might have been improved with less onerous data collection.
Primary effectiveness results and sample size calculations
The alternative primary effectiveness outcome was used for further analysis as described above. Forty-five of a possible 49 pain scores were recorded across both arms (table 3). Patients in the treatment arm appeared to experience less pain compared with the control arm (ordinal logistic regression OR 0.495, 95% CI 0.158 to 1.554). It was estimated that 207 patients would be required to achieve 90% power at the 5% level to detect a difference in pain similar to this feasibility study, allowing for 10% loss of primary outcome data.
Distribution of the alternative primary outcome by arm in the 45 patients providing data
Qualitative interviews with patients in the treatment arm suggested they valued the localised and continuous nature of the PNC, feeling reassured that their pain would be managed well.
Other effectiveness results
Multiple secondary effectiveness results were analysed. Details of treatment failure, CSP, PLP, SSI, nausea and vomiting, S-LANNS, opioid use, OBAS, anxiety (from HADS) and depression (from HADS) are given in tables 4 and 5. Significant missing data at follow-up resulted in wide CIs for estimated effect sizes. Nerve identification and successful placement of the PNC was 100%.
Results of logistic regression on dichotomous secondary outcomes (post-surgery, discharge, month 2 and month 6)
Descriptives and results of non-parametric Mann-Whitney U (MWU) tests or linear regression on continuous secondary outcomes (post-surgery, day 5, month 2 and month 6)
Trial acceptability and understanding
Qualitative interview data indicated that patients found trial processes acceptable and were happy with the randomisation aspect of the trial. Healthcare professionals reported that patients who consented to take part understood the trial information provided, and that written trial information was clear and of a suitable level. Despite this, the majority of patients did not demonstrate a good understanding of the trial at the time of interview, particularly randomisation processes. Interviewees’ ability to give an accurate account of trial information may have been affected by difficulty recalling information given preoperatively rather than a lack of understanding. However, thoroughly checking patient understanding at the time of consent will be an important consideration for future trials in this area.
Some healthcare professionals felt that it was preferable from a pain management perspective for patients to be in the intervention arm. Therefore, reassurance that pain will be managed effectively for these patients would be key for future trials to ensure engagement and equipoise.
Health economic analysis
The costs associated with the PNC were estimated at £130 per participant (online supplementary table 3). Mean length of postoperative stay was approximately equivalent between the two arms (inpatient stay until medically fit for discharge: treatment arm (mean±SD) 24.43±21.28 days; usual care: 24.05±18.46 days). Based on available cases the pattern of healthcare resource use over 6 months was similar for both groups, with hospital admissions (elective and non-elective), out-patient attendance and primary care (district nurse and general practitioner consultations) being the key drivers of resource use (online supplementary table 4). Resource use data were highly skewed, with baseline differences between the two arms evident. Similar to the issues of attrition identified for collection of other patient-reported data at follow-up above (table 2), there was evidence of significant missing data for the patient-reported resource use questionnaire. Limited data obtained on direct and indirect costs to the patient and family precluded formal examination.
Discussion
The PLACEMENT trial has demonstrated that recruitment into a randomised controlled trial comparing PNC use to standard care was feasible. Recruitment rates were excellent, with over 60% of all eligible patients being recruited. Patients and healthcare professionals found trial processes acceptable. Nerve identification and PNC placement rates were 100%. Using the alternative primary outcome, over 90% of patients provided enough primary outcome data. Attrition rates were very high at 2 and 6 month follow-up and need to be accounted for in any future effectiveness trial.
Patients undergoing amputation for PVD are often comorbid and suffer with cognitive impairment.37 It is unsurprising that the most frequent reason for ineligibility at screening is due to a lack of mental capacity. Of the 76 eligible patients, 49 were recruited. This proportion (64.5%) is in line with the results of a recent review of 151 Health Technology Assessment (HTA) funded RCTs, reporting a proportion of 70% (IQR 51% to 87%) of successfully completed trails.38
We chose the 11-point VRS as the tool to provide our principle primary outcome as it is recommended by multiple studies of pain measurement.31 During trial set-up, we identified it may be difficult to capture a significant number of measurements using this scale, which is why we also recorded routinely collected 4-point VRS measurements. Only 69.4% of patients provided principle primary outcome data, whereas over 90% of patients supplied alternative primary outcome data, which used the routinely collected 4-point VRS score. Admittedly this is using an arbitrarily definition of ‘sufficient data’ (greater that 50% completion rate), as defined by the TMG. However, this is in line with outcome data rates from the HTA review of successful trials (89%, IQR: 79% to 97%), supporting use of the alternative primary outcome in a future effectiveness trial.38 Any future trial should aim to use whichever pain scale is routinely collected in the trial centre, with all data then converted to the 4-point scale for analysis.
Attrition rates at follow-up were markedly high. It is well recognised that amputees suffer high rates of mortality, with figures from some studies as high as 50% at 1 year. While death accounted for only eight patients (16.3%) at 6 months, several other patients were hospitalised for other medical problems, still in rehabilitation centres, or otherwise uncontactable. While not necessarily unsurprising, the difficulty with obtaining patient-reported data at 2 and 6 month follow-up limit the ability to investigate the effect of postoperative PNC usage on PLP and CSP. There have been relatively few randomised trials recruiting amputees, and the long-term attrition rates found here will be important to consider in any future trials.
Assessing postoperative pain is not standardised or straightforward. No clear guidelines exist for specifying which tool, at what frequency should be used nor how it should be analysed. It is also not clear how best to adjust for baseline/preoperative pain. A systematic review published in 2011 identified 54 papers comparing different tools for assessing postoperative pain, of which 52 used Visual Analogue Scale, 39 used VRS and 32 a numerical rating scale.39 Use of a numerical rating scale had better compliance in the majority of studies reporting this, and was the recommended tool in 11 of the studies. A more recent systematic review of postoperative pain reporting after regional analgesia identified 15 different outcome measures used in 31 different studies, with some including pain on movement as well as at rest.40 Five studies recorded maximal pain score during the study period. This heterogeneity in reporting is a problem in many disciplines, and the COMET (Core Outcome Measures in Effectiveness Trials) group aims to standardise reporting of outcome measures. While a core outcome set for chronic pain has been published, none exist for postoperative pain.31
A number of tools were used to assess pain other than the VRS. The S-LANNS scores were captured at day 0 and 5, and at 2 and 6 months. S-LANSS is a tool predominantly used for differentiating between nociceptive and neuropathic pain in patients with chronic pain, and its use and value in the immediate postoperative period is uncertain. Completion rates were approximately 84%, so it may be useful to identify pain type, but whether this is of prognostic value remains unknown. The OBAS tool was developed for use in the postoperative period and attempts to capture a quantitative measure of overall patient satisfaction with analgesia. While the composite score contains a rating for current pain experience, it is heavily weighted towards the side effects of opioid analgesia. Completion rates were reasonable, but it lacked discriminatory power in this study, with minimal between-group differences. Long-term CSP and PLP were assessed by the Groningen questionnaire, although these data were subsequently dichotomised using an arbitrarily decided division point. This approach makes the data easier to quantify and interpret, but fails to account for degree of pain, or episodic but very severe pain,
Addressing the cost-effectiveness of PNC as a secondary objective is feasible in a future effectiveness trial, but a number of design issues would need to be considered. The challenge of obtaining resource use data could be resolved by capturing this information as part of the clinical trial record, with key drivers such as hospital admission collected from routine records. While the EQ-5D is a well-established measure to derive utilities, the challenges of baseline imbalance, missing data and lack of sensitivity are important considerations for a future trial-based economic analysis. Postoperative PNC use is, by definition, a short-term intervention, and longer-term QoL-related utility differences are unlikely to be large. Economic evaluation of PNC use, costing only £130 to administer, should most likely focus on demonstrating non-inferiority rather than superiority in an effectiveness study.
Qualitative interviews proved an invaluable addition to the feasibility process. The difficulty in deciding on a pain assessment tool was noted by the TMG. While no clear consensus emerged in our interviews with patients and healthcare professionals as to which tool was preferred, it was noted that using more than one pain scale was confusing for patients. Future trials should therefore consider using whichever pain tool is already used at their institution. Interviews also identified a lack of patient understanding of the trial. Our experience is that patients undergoing amputation often have mild cognitive impairment, and we suggest this finding may represent a lack of patient recall, rather than inadequate consent technique. Issues around patient understanding of the informed consent process have been reported previously. For example, the systematic of Falagas et al 41 found that adequate understanding of the randomisation process by patients was demonstrated in only 50% of the studies providing relevant data. Any future efficacy trial would need to take account of this specific issue and, after careful discussion with patients, decide on approaches to maximise patient understanding and retention of trial information. This may include the use of interventions with evidence of efficacy,42 including multimedia content of the trial process, and an extended discussion for consent.
One potential limitation of this study is that it did not assess the use of a placebo arm, with infusion of a non-active substance. ‘Soft’ outcomes such as pain scores are known to be susceptible to the placebo effect, so any benefit seen in the treatment arm could be a result of this.43 However it could be argued that even if the effect of the perineural catheter is not mediated by the local anaesthetic nerve blockage, if it is shown to be of benefit over usual care then this is a result which is likely to translate into clinical practice, so is still of value. We therefore feel that the pragmatic design of simply comparing placement and use of the PNC to usual care is appropriate. Placebo interventions in surgical trials are also well recognised as being difficult, and face ethical issues over and above medical trials.44
Conclusion
This study has shown that recruitment to an RCT evaluating the effectiveness of a PNC on postoperative pain in amputees is feasible, with recruitment rates comparable to recently completed efficacy trials. Immediate postoperative pain was more efficiently captured using a 4-point VRS than with an 11-point VRS. Opioid consumption is also well captured. Significant attrition should be expected and planned for, which could limit investigation into the effects of PNC usage on PLP and CSP.
Acknowledgments
In addition to the authors, the PLACEMENT team comprises Judith Evans, Melanie Morgan, Heather Strange and Sam Clarkstone. The authors would like to acknowledge the contribution of David Cox and Sian Jones, as the patient and public representatives on the Trial Management Group. The authors would also like to acknowledge the contribution of the Trial Steering Committee members, namely Paul Hayes, Nia Goulden, John Bevan and Geinor Bean. The study would not have been possible without the support of the vascular and research nursing teams at the two sites including David Lewis, David Mclain, James Tozer, Edward Brown, Kristian Glover, Heeam Nassa, Annie Clothier, Tracey Hutchings and Elaine Wall at the Royal Gwent Hospital, Newport; and Chris Davies, Amy Stimpson, Kamran Mohiuddin, Andrew Beamish, Luke Hopkins, the Health and Care Research Wales Workforce, the South West Wales Research Network, Sharon Storton, Leanne Quinn, Marie Williams, Lynda Connor and Glyn Gainard at Morriston Hospital, Swansea.
References
Footnotes
Twitter @davebosanquet, @GraemeAmbler
DCB and GKA contributed equally.
Correction notice This article has been corrected since it was published. Author initials have been updated.
Contributors CPT and DCB were co-Chief Investigators of this study. CPT, DCB and GKA led the development of the research question, study design, obtaining funding, obtaining ethical and regulatory approval and implementation of the study protocol, along with ET-J, MK, LB-H, and DF. CAW was the Trial Manager and ET-J the Senior Trial Manager who coordinated the operational delivery of the study protocol and recruitment. LB-H and SM undertook patient and staff interviews and data interpretation of the qualitative part of the study. DF was the Health Economist and undertook the economic evaluation with GKA and TP. DH was responsible for data management. MK provided expert statistical advice during development of the study. TP developed the statistical analysis plan with input from GKA, provided expert statistical advice during the recruitment phase of the study and undertook the final statistical analysis. NS provided expertise in pain management. All authors listed provided critical review and final approval of the manuscript. Neither the sponsor nor funders have been or will be involved in or have authority over study design, management, analysis and interpretation of data, writing of the report or decision to submit the report for publication.
Funding This project was funded by the Welsh Government through Health and Care Research Wales. This was through the Patient and Public Benefit (RfPPB) scheme, reference number 1198, and was sponsored by Aneurin Bevan University Health Board, Newport, South Wales.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Full ethical approval has been granted (Wales REC 3 reference number 16/ WA/0353). Written informed consent was obtained from all participants in the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The quantitative datasets generated during the study can be made available upon request from opendata@cardiff.ac.uk. Data are generally freely available, but recipients are expected to acknowledge the original creators in any public use of the data or in publishing research results based wholly or in part upon the data – anyone requesting access to data will be asked to agree to the terms of the Creative Commons Attribution 4.0 license. The trialists may ask the requestor to cover reasonable cost for preparing and providing the data (for example physical storage and postage, where dataset size makes it impractical to provide data by electronic means).