Article Text

Abstract
Introduction An ongoing outbreak of yellow fever (YF) has been reported in Brazil with 1261 confirmed cases and 409 deaths since July 2017. To date, there is no specific treatment available for YF. Recently published papers describing in vitro and animal models suggest a potential effect of antiviral drugs (approved for the treatment of hepatitis virus) against flaviviruses, including YF. The primary aim of this study is to analyse the effect of sofosbuvir on viral kinetics and clinical outcomes among patients presenting with YF. This is a multicentre open-label randomised controlled trial with 1:1 individual allocation, stratified by severity and by recruiting centre.
Methods and analysis Adults with suspected or confirmed YF infection and symptoms lasting up to 15 days are screened. Eligible and consenting patients are randomised to receive oral sofosbuvir 400 mg daily for 10 days or to receive standard clinical care. Viral kinetics are measured daily and the reduction in YF plasma viral load from the sample at inclusion to 72 hours after randomisation will be compared between active and control groups. Clinical outcomes include severity meeting criteria for intensive care support, liver transplantation, in-hospital mortality and mortality within 60 days.
Ethics and dissemination Ethics approval was obtained at the participating sites and at the national research ethics committee (CAAE 82673018.6.1001.0068). The trial has been submitted for ethical approval at additional potential recruiting centres. Results of the study will be published in journals and presented at scientific meetings.
Trial registration Brazilian Clinical Trials Registry (RBR-93dp9n).
- yellow fever
- sofosbuvir
- randomised controlled trial
- viral kinetics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
In this open-label 1:1 parallel group randomised controlled trial, we will address the effect of oral sofosbuvir at 400 mg daily dose compared with standard clinical care for patients with yellow fever (YF) infection referred for hospitalisation during a YF outbreak in Brazil.
Randomisation will be done using an electronic platform, stratified by clinical severity and recruiting centre.
Median change in plasma YF viral levels at 72 hours after inclusion and clinical outcomes (meeting severity criteria for intensive care support, liver transplantation, in-hospital mortality, mortality within 60 days) will be compared between active and control groups.
An independent data safety monitoring group will be designated to supervise severe adverse events related to the study medication and to perform an interim analysis after the inclusion of 2/3 of predicted total sample size.
Introduction
There is an ongoing outbreak of yellow fever (YF) in Brazil, with an increasing number of cases since 2016, notably in the Southern region. Between July 2017 and May 2018, 1261 confirmed YF cases were reported, with 409 deaths (mortality rate of 32.4%). Nowadays, a major part of population at risk for YF resides in newly affected areas (58.3%), where vaccination was not previously recommended.1
YF has a wide clinical spectrum, from asymptomatic to severe haemorrhagic disease associated with liver and renal failure and high case fatality rate. Vaccine safety remains a concern, especially because the yellow fever vaccine-associated viscerotropic disease (YFV-AVD) is a rare condition but has a high case-fatality rate. To date, there is no specific treatment available for YF or YFV-AVD.2
Recently published papers describing in vitro and animal models suggest a potential effect of sofosbuvir (an antiviral approved for the treatment of hepatitis C virus) for other flaviviruses, such as dengue, zika and YF.3–6
A group of researchers showed that sofosbuvir inhibited YF virus replication in different lineages of human hepatoma cells and reduced the YF-induced mortality and lack of weight gain in neonatal mice and suggested that sofosbuvir should be considered for clinical use in YF-infected individuals.5
Another research team tested the in vitro activity against YF and zika virus of several antivirals used for hepatitis C virus (ribavirin, daclastavir, sofosbuvir and ledispavir/sofosbuvir combination). All the tested drugs presented activity and selectivity against YF and zika virus in human hepatoma cells, but ledispavir/sofosbuvir combination and sofosbuvir showed the best antiviral activity towards both viruses.7
Although there is some evidence of sofosbuvir activity against YF, there are no data from human studies as to the effect of sofosbuvir on acute YF.
We present the protocol for a multicentre open-label randomised controlled trial to analyse the effect of sofosbuvir on YF viral kinetics and on clinical outcomes (severity meeting criteria for intensive support, liver transplantation, in-hospital mortality and mortality within 60 days among patients presenting with acute YF). This is a multicentre open-label randomised controlled trial with 1:1 individual allocation, stratified by severity and by recruiting centre.
Methods and analysis
Study setting
The study will be conducted in secondary and tertiary healthcare units in Brazil. At this moment two recruiting centres are actively enrolling, both of which are tertiary teaching hospitals in São Paulo city (São Paulo State, Brazil). Six additional recruiting centres in the States of São Paulo and Minas Gerais are waiting for protocol ethical approval. The inclusion of participants has begun in March 2018 and the protocol is still in recruiting phase.
Eligibility criteria
Adults≥18 years of age hospitalised at the recruiting centres with suspected or confirmed YF infection and with symptoms lasting up to 15 days are being screened.
We are including patients with a history of fever (axillary temperature above 37.8°C), exposed to geographic areas with risk for YF transmission (according to updated epidemiological reports) and with transaminases (either alanine aminotransferase or aspartate aminotransferase) above 500 U/L.
We are excluding patients with amiodarone use in the last 72 hours, current pregnancy, women refusing to interrupt breastfeeding, current use of sofosbuvir for hepatitis C treatment, those already tested with a negative plasma YF real-time polymerase chain reaction (RT-PCR) prior to inclusion and those with another known aetiology that justified all signs and symptoms.
Interventions
Eligible participants are randomised to receive either sofosbuvir 400 mg daily for 10 days or standard care. Patients should remain hospitalised for at least 10 days after the randomisation.
For those allocated to receive sofosbuvir, the oral route is preferred, although enteral tube administration is also acceptable.
The following criteria determine immediate interruption of sofosbuvir prescription: negative YF RT-PCR in a test performed at enrolment; amiodarone prescription; anaphylactic shock or grade III rash attributed to sofosbuvir; inability to receive oral or enteral tube medications.
In the case of sofosbuvir interruption, patients will continue to be followed-up for adverse events (AE) and outcomes according to the protocol.
In case of withdrawal of consent, the study medication and study procedures will be interrupted, but the participant will be asked to undergo safety evaluations according to the protocol if possible.
Outcomes
YF plasma viral load will be measured daily and median change in plasma YF viral levels at 72 hours after inclusion will be compared between active and control groups.
The YF RT-PCR assay employed has a sensitivity of 17 000 copies/mL.
Clinical outcomes (severity meeting criteria for intensive support, liver transplantation, in-hospital mortality, mortality in 60 days) will also be compared between active and control groups. Time to YF RT-PCR clearance in biological samples and overall survival will also be evaluated.
Criteria for intensive support is defined by any of the following conditions: transaminases>3000 U/L; international normalised ratio >1.5; platelet counts >90 000/mm³; renal impairment (creatinine >1.2 mg/dL or >50% increase in baseline creatinine); bleeding; encephalopathy ≥grade I8 or seizures; mechanical ventilation; or hypotension requiring use of vasoconstrictive drugs.
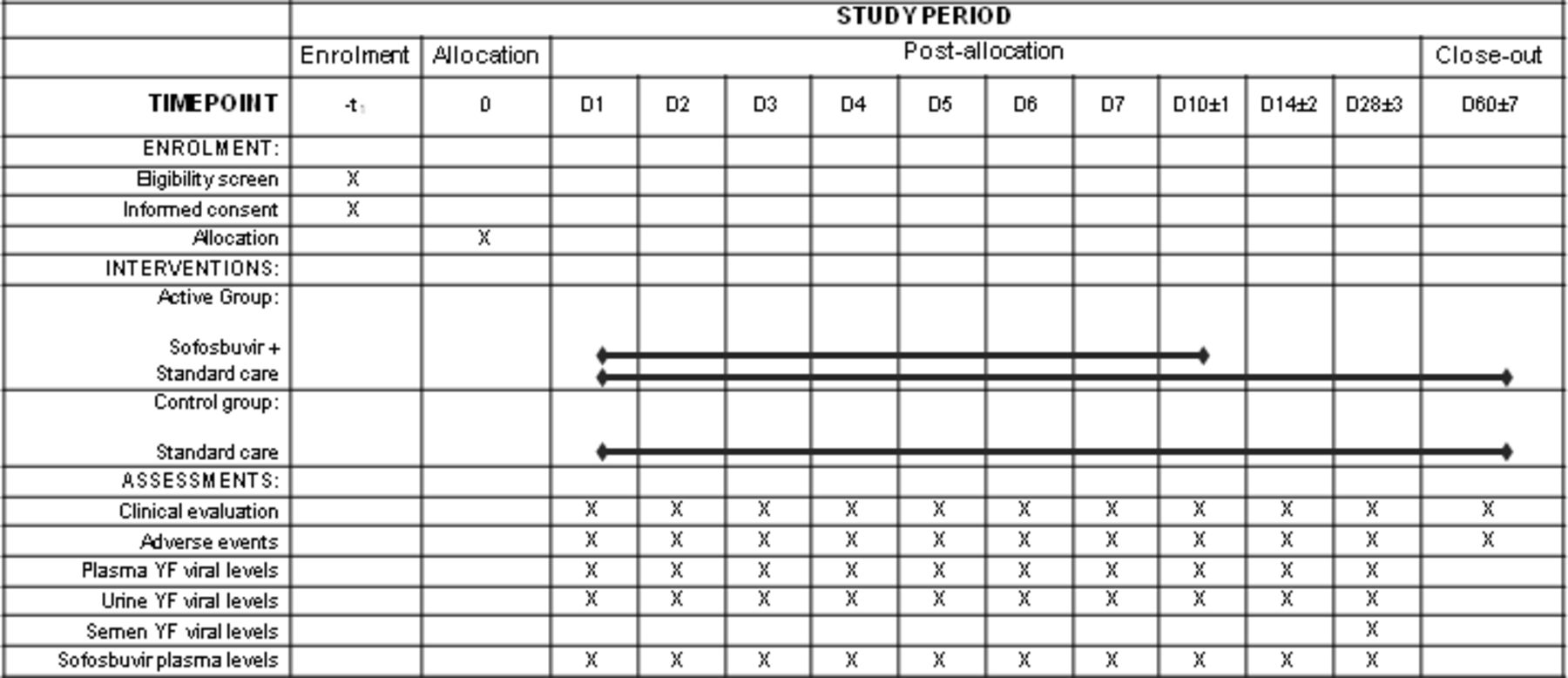
The timeline of procedures is illustrated in figure 1.
{kind=link}
Timeline of procedures of SOFFA study. YF: yellow fever. D1 corresponds to the allocation moment until 23h59; D2: from 0h until 23h59, D3: from 0h until 23h59 and so on. Clinical evaluation and adverse events assessments on D60 could be done by phone contact.
Sample size
The sample size calculation considered the estimated effect of sofosbuvir in YF-infected patients submitted to the compassionate use of this drug.7
Our primary outcome, median change in plasma YF viral levels at 72 hours after inclusion in the study, will be indirectly measured by the cycle threshold value (Ct) of the RT-PCR.
Assuming a two-sided 5% significance level and a power of 80%, we estimated the sample size per group with different estimates of drug effect and dispersion (SD) of the variable, as shown in table 1.
Sample size based on different estimates of difference between groups (measured by increase in the cycle threshold value at 72 hours after inclusion) and variant estimates of SD
Considering the presented estimates, the calculated sample size was defined as 90 participants (45 per group).
Recruitment
All consecutive eligible patients hospitalised in the participating centres with confirmed or suspected acute YF will be screened. The investigators will contact the hospital epidemiology surveillance team on a daily basis to capture all confirmed or suspected acute YF cases admitted in any unit of the hospital over the course of the study.
Assignment of interventions
The random allocation sequences were generated by one investigator (not involved in participant screening or enrolment) using the electronic platform Sealed Envelope.9 Allocation was created in a 1:1 proportion using permuted blocks of size four or six and stratified by recruiting centre and severity (moderate or severe strata).
The sequence was transferred to sequentially numbered, opaque, sealed envelopes which are maintained in locked cabinets accessible only to investigators performing screening and randomisation procedures.
Participants are randomised for severe stratum if they meet any criteria for intensive support.
Patients and healthcare providers will be not be blinded regarding study allocation, but the intervention will not be disclosed to laboratory personnel running YF RT-PCR tests during the study.
Data collection methods
Clinical information and medical history will be obtained at enrolment directly from the participant, family members or medical charts. Laboratory test results will be retrieved from medical charts and laboratory reports.
Dialysis requirement will be assessed daily by the attending physicians and the evaluated criteria such as creatinine levels, urine output, metabolic acidosis, catecholamine infusion, mechanical ventilation encephalopathy grade and active bleeding will be recorded, as well as the receipt of renal replacement therapy.
Plasma concentrations of sofosbuvir will be determined in blood samples obtained daily during administration of sofosbuvir using liquid chromatography-tandem mass spectrometry (with a lower limit of quantification of 1 ng/mL).10 11
All study variables will be collected and managed using REDCap electronic data capture tools hosted at the Faculdade de Medicina da Universidade de Sao Paulo (FMUSP).12
Data management
The electronic data collection tools will be developed using appropriate range checks and validation rules. Investigators will obtain access to the electronic forms using password-secure unique usernames. Internal data monitoring will be conducted during the trial for cross-validation of collected information.
Statistical methods
The main analysis in the study will be based on the intention-to-treat principle. However, we will also include per-protocol analysis and comparisons stratified by severity at inclusion. Demographic and clinical characteristics will be presented using frequencies, means/medians, SD and interquartile ranges with their corresponding 95% CIs. Comparisons between groups will be made using the χ2 test or Fisher’s exact test for categorical variables and T-test or Wilcoxon’s rank-sum test for continuous variables as appropriate. The independent effect of sofosbuvir on the endpoints of viral kinetics and of clinical outcomes will be analysed through multivariate models adjusted for the potential confounding factors not controlled by random allocation. The effect of sofosbuvir on the time to achieve undetectable YF RT-PCR in biological specimens will be analysed using survival curves and Cox’s proportional regression models adjusted for potential confounders.
Data monitoring
An independent Data Safety Monitoring Board (DSMB) was designated to oversee the safety and welfare of study participants and to provide recommendations based on reported AE. It will evaluate the interim analysis regarding whether the study should continue without change, be modified, or be terminated. The DSMB is composed of three consultants including one infectious diseases specialist, one intensive care physician and one epidemiologist. An interim analysis will be conducted after the inclusion of the first 60 participants. The DSMB will meet at the beginning of the study to determine specific methods for interim analysis and stopping guidelines.
Harms
Solicited and spontaneous AE will be monitored over the course of the study. Serious AE (SAE) will be reported to the DSMB and to the local Institutional Review Board (IRB) as soon as identified by the study investigators. In addition, a report describing non-serious AE grade≥3 will be provided bimonthly to the DSMB and to the IRB. Additional information about the reported AEs, including the outcomes related to each AE, will be forwarded as soon as available.
We will consider as SAE those: resulting in death, representing life threat, requesting hospitalisation or extending current hospitalisation, resulting in persistent or significant disability, resulting in congenital abnormality or birth defect, or including important medical events, even if they do not represent life threat or hospitalisation, but that may be risky to the patient or may require intervention to prevent one or more of the events listed above.
Patient and public involvement
There was no involvement of patients or public in the conception or conduct of this study. At any time, participants can be informed about study outcomes through the principal investigator.
Ethics and dissemination
This study will be conducted according to the Good Clinical Practices of the International Conference on Harmonization Good Clinical Practices (ICH-GCP), and to local and national regulation. Results of the study will be published in journals and presented at scientific meetings.
Written informed consent will be obtained for each participant. We will invite the patient directly to enrol in the study if possible, and a legal representative or family will be contacted to take part in the informed consent process if necessary.
Personal information about potential and enrolled participants will be collected and shared only through RedCap platform,12 accessible only by authorised trial investigators. Personal identifier information will be linked to stored data or samples solely by a protected master list kept under lock locally and will not be shared outside the study staff at the local partner site during and after the trial.
We used the SPIRIT checklist when writing this manuscript.13
The final trial dataset will be available only to investigators responsible for analysing the data.
Dissemination policy
Trial results will be communicated to healthcare professionals, the public and other relevant groups via publication, reporting in results databases or other data-sharing arrangements as appropriate. Communication of trial results to participants will be done if direct benefit or prevention of harm is anticipated from disclosure of the study results. Study sponsors will have no role in study design, data collection, analysis, interpretation of results, manuscript writing or decision to publish resulting reports.
Acknowledgments
We are thankful to Marjorie Shafferman for her review of the manuscript.
Footnotes
Twitter @luciana_vb, @jrrpinho
Contributors CF-M, LVBC, VIA-S, YL-H, JS, DJ, MBA, JRRP, FdMM, MSGG, APMS, APC, CHVM, AFR, ACdSSN, CMSM, RFAT, LMSB, MPG, LCPJ, TNLS, ATWS, LACA, EA, WA, RBdM, LD, GMA, LMSM, IMdS, FJC, ECS and ASL are the authors. CFM, LVBC, VIA-S, YL-H, JS, JRRP, FdMM, MSGG, APMS, APC, CHVM, AFR, ACdSSN, CMSM, RFAT, MPG, LCPJ, TNLS, ATWS, FJC, ECS and ASL conceived the idea of study and its design. CFM, LVBC, YL-H, JS, DJ, MBA, CHVM, AFR, ACdSSN, CMSM, RFAT, LMSB, ATWS, LACA, EA, WA, RBdM, LD, GMA, LMSM, IMdS are essential to data collection. CFM, LVBC and VIA-S wrote the first draft of the manuscript. ECS and ASL provided advice and supervision. All authors met the criteria: (1) Substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work. (2) Drafting the work or revising it critically for important intellectual content. (3) Final approval of the version to be published. (4) Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This work was supported by FAPESP, São Paulo Foundation for Research [grant number 2016/01735-2] and Fundação Faculdade de Medicina USP. The funders had no role in study design, data collection and analysis, decision to publish, or in the preparation of the manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Ethics approval was obtained at the participating sites (Comissão de Ética para Análise de Projetos de Pesquisa do Hospital das Clínicas da FMUSP and Comitê de Ética em Pesquisa do Instituto de Infectologia Emílio Ribas) and at the national research ethics committee (CAAE 82673018.6.1001.0068). The trial is currently under evaluation by the research ethics committee at additional potential recruiting centres.
Provenance and peer review Not commissioned; externally peer reviewed.