Article Text

Abstract
Introduction Fibromyalgia (FM) is a common debilitating condition with limited therapeutic options. Medications have low efficacy and are often associated with adverse effects. Given that FM is associated with a defective endogenous pain control system and central sensitisation, combining interventions such as transcranial direct current stimulation (tDCS) and aerobic exercise (AE) to modulate pain-processing circuits may enhance pain control.
Methods and analysis A prospective, randomised (1:1:1:1), placebo-controlled, double-blind, factorial clinical trial will test the hypothesis that optimised tDCS (16 anodal tDCS sessions combined with AE) can restore of the pain endogenous control system. Participants with FM (n=148) will undergo a conditioning exercise period and be randomly allocated to one of four groups: (1) active tDCS and AE, (2) sham tDCS and AE, (3) active tDCS and non-aerobic exercise (nAE) or (4) sham tDCS and nAE. Pain inhibitory activity will be assessed using conditioned pain modulation (CPM) and temporal slow pain summation (TSPS)—primary outcomes. Secondary outcomes will include the following assessments: Transcranial magnetic stimulation and electroencephalography as cortical markers of pain inhibitory control and thalamocortical circuits; secondary clinical outcomes on pain, FM, quality of life, sleep and depression. Finally, the relationship between the two main mechanistic targets in this study—CPM and TSPS—and changes in secondary clinical outcomes will be tested. The change in the primary efficacy endpoint, CPM and TSPS, from baseline to week 4 of stimulation will be tested with a mixed linear model and adjusted for important demographic variables.
Ethics and dissemination This study obeys the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of Partners Healthcare under the protocol number 2017P002524. Informed consent will be obtained from participants. Study findings will be reported in conferences and peer-reviewed journal publications.
Trial registration number NCT03371225.
- transcranial direct current stimulation
- aerobic exercise
- fibromyalgia
- endogenous pain control system
- temporal slow pain summation
- conditioned pain modulation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- transcranial direct current stimulation
- aerobic exercise
- fibromyalgia
- endogenous pain control system
- temporal slow pain summation
- conditioned pain modulation
Strengths and limitations of this study
A sham-controlled, powered clinical trial on a novel low-cost therapy for fibromyalgia.
Endogenous pain system biomarkers will help reveal the mechanisms of fibromyalgia as well as the interventions.
This study will inform us on the number of sessions needed to induce significant changes in neuroplasticity reflected in the above mentioned markers.
The secondary outcomes of this study will evaluate the suitability of the proposed biomarkers to predict treatment response.
Exclusion of patients with increased risk during exercise may limit the generalisability of the findings.
Introduction
Fibromyalgia (FM) affects about 2% of the world population1 and is associated with poor quality of life mainly due to pain, fatigue, sleep disturbances, functional limitations and cognitive impairments.2 Current treatments for this challenging complex condition lead to an average annual cost of $5945 in insurance claims per patient with FM, more than twice the amount of a typical beneficiary.3 The treatment of choice is a multimodal approach that includes self-management strategies,4 but there is a large gap between supply and demand as access to such therapies is limited. Consequently, many patients with FM rely on pharmaceuticals such as non-steroidal anti-inflammatory drugs, antidepressants and/or anticonvulsants, which usually do not provide enough symptom relief and are frequently associated with adverse effects.5 Therefore, there is an urgent need for the development of novel and targeted treatments with fewer side-effects.
Rationale and gap
Accumulating evidence6–9 shows that disturbances in the endogenous pain control system lead to chronic pain. Several neurophysiological10–16 and neuroimaging17–21 studies showed altered pain processing mechanisms in FM; therefore, therapies that target and modulate the neural circuits involved in pain control are essential to treat FM characteristic chronic widespread pain. Different ways to potentially modulate these circuits include exercise—which has a known evidence-based therapeutic effect on pain in FM,22 and non-invasive neuromodulation techniques such as transcranial direct current stimulation (tDCS)—which demonstrably improve several chronic pain conditions.23–28 Despite its investigated benefits to treat different pain conditions (typically targeting the primary motor cortex (M1)), tDCS effects in FM have been mixed.29–32 Yet, tDCS can be easily coupled to other therapies due to its low-cost and portability,33 and such combinations have been superior to either of the therapies alone in other disorders.34–36 We have shown in a pilot study with 45 FM subjects that combining exercise and tDCS for FM leads to a significant pain decrease that also shows a different neural signature as compared with each therapy alone (tDCS or exercises).37 In this initial study, however, the endogenous pain inhibitory system was not assessed.
Given the extensive data showing that (i) FM has a defective endogenous pain inhibitory system10–16 and (ii) exercises38–40 and tDCS lead to modulation of this system,31 41 42 we then hypothesised that these two neuromodulatory techniques can help restore the endogenous pain inhibitory system in FM. Neurophysiological and clinical assessments including electroencephalography (EEG), transcranial magnetic stimulation (TMS), quantitative sensory testing and questionnaires for pain and quality of life can provide important data to understand how the endogenous pain inhibitory system is then modulated by these two interventions.
Research question and hypothesis
We therefore aimed to test whether in subjects with FM 16 sessions of M1 anodal tDCS combined with aerobic exercise (AE) decrease temporal slow pain summation (TSPS) and increase conditioned pain modulation (CPM) responses compared with each intervention alone and to sham when assessed on the last day of intervention. We hypothesise that this optimised tDCS plus AE technique will lead to a stronger engagement of the endogenous pain regulatory system, which will ultimately lead to increased pain regulation in patients with FM.
Objectives
Primary objective
To evaluate the effects of 4 weeks of tDCS plus AE on the endogenous pain regulatory system (assessed by CPM) and central sensitisation (assessed by TSPS) compared to either interventions alone and to no intervention.
Secondary objectives
To determine the effect of these interventions on cortical markers of inhibitory control that are also altered in FM, such as intracortical inhibition assessed by TMS, and changes in thalamocortical dysrhythmia (TCD) and event-related desynchronisation (ERD) assessed by EEG.
To assess whether engagement of the two main targets tested in this study (TSPS and CPM) are associated with the secondary clinical outcomes (i.e., changes in pain outcomes: Brief Pain Inventory, Revised Fibromyalgia Impact Questionnaire).
To assess EEG changes across groups and their suitability as potential markers of TCD normalisation.
To determine the number of sessions needed to induce significant changes in markers of the endogenous pain inhibitory system and central sensitisation (CPM and TSPS) and cortical changes (paired pulse TMS and EEG).
Methods and analysis
Trial design
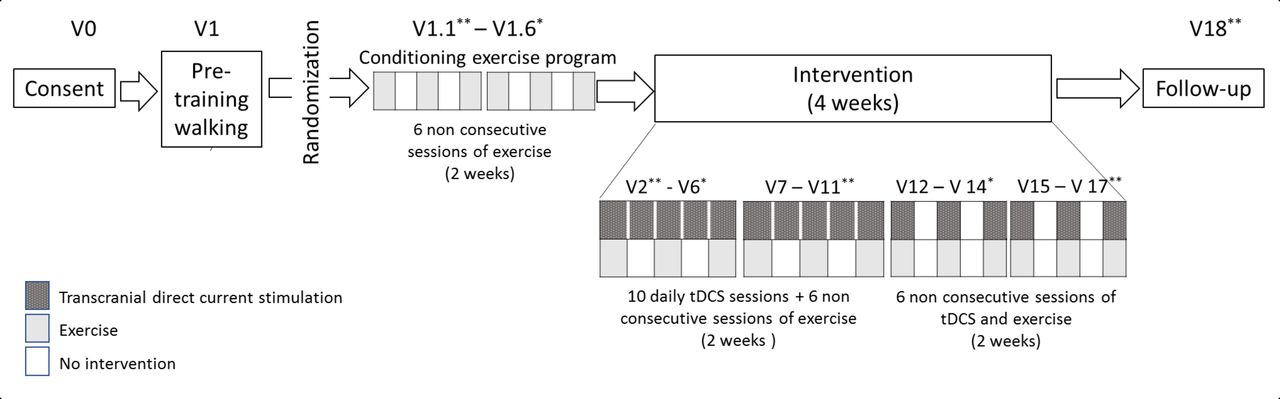
This is a single centre 4-arm factorial Randomized Clinical Trial (RCT). Participants will be randomised using a random blocked randomisation sequence generated by a computer software. We used a 1:1:1:1 allocation ratio to active or sham tDCS combined with AE or non-aerobic exercise (nAE) on the first day of the conditioning exercise programme. The staff member performing randomisation will not be involved in the trial otherwise. Sequentially numbered sealed envelopes will maintain allocation concealment. Investigators providing assessments will be blinded to tDCS but not exercise. Assessors of primary and secondary outcomes (and participants) will be blinded to group allocation (see figure 1 for group allocation).

Flowchart of the study based on CONSORT criteria. AE, aerobic exercise; nAE, non-aerobic exercise; tDCS, transcranial direct current stimulation.
Study setting
This is a single-site study, and all procedures will be conducted at the Neuromodulation Center, Spaulding Rehabilitation Hospital. Enrolment start date is 1 May 2019 and expected end date is 31 December 2023.
Eligibility criteria
We will use broad-based recruitment strategies, including online advertisements, flyers, clinician referrals and so on. All eligible participants must fulfil the inclusion criteria and have none of the exclusion criteria listed in box 1.
Inclusion and exclusion criteria
Inclusion criteria
Age range 18–65 years.
Diagnosis of fibromyalgia pain according to the American College of Rheumatology (ACR) 2010 criteria (existing pain for more than 6 months with an average of at least 4 on a 0–10 Visual Analogue Scale (VAS) scale) without other comorbid chronic pain diagnosis.
Pain resistant to common analgesics and medications for chronic pain such as Tylenol, Aspirin, Ibuprofen, Soma, Parafon Forte DCS, Zanaflex and Codeine.
Must have the ability to feel sensation by Von-Frey fibre on the forearm.
Able to provide informed consent to participate in the study.
Exclusion criteria
Clinically significant or unstable medical or psychiatric disorder.
History of substance abuse within the past 6 months as self-reported (if subject reports a history of substance abuse, we will confirm using the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM V criteria).
Previous significant neurological history (eg, traumatic brain injury), resulting in neurological deficits, such as cognitive or motor deficits, as self-reported.
Previous neurosurgical procedure with craniotomy.
Severe depression (with a score of >30 on the Beck Depression Inventory).
Pregnancy—as the safety of tDCS in pregnant population (and children) has not been assessed (though the risk is non-significant), we will exclude pregnant women (and children). Women of childbearing age will be required to take a urine pregnancy test during the screening process and in every week of stimulation).
Current opiate use in large doses (more than 30 mg of oxycodone/hydrocodone or 7.5 mg of hydromorphone (Dilaudid) or equivalent).
Patients will be excluded when they have increased risk for exercise defined as (i) not fulfilling the American College of Sports Medicine criteria (ie, risk of cardiovascular complication48) and in this case not cleared by a licensed physician.
As part of the eligibility criteria, participants will perform a pretraining visit to evaluate if they are comfortable with walking on the treadmill at a self-selected speed at their baseline heart rate (HR) for 30 min. Only subjects comfortable with this task will be randomised. If the subject is unable to walk for 30 min on the treadmill or reports discomfort or any side-effects precluding physical exercise (eg, excessive muscle soreness), they will be screened out. Also, a demographic survey will be taken during the consent visit.
Intervention
Exercise
Conditioning exercise programme: 6 exercise sessions are divided in 3 days per week over 2 weeks. Duration of sessions will start with 10 min and increase gradually, ending with a 30 min session on the last day. The AE group will walk briskly at 60%–70% of their maximum HR and the nAE group will walk within 5% of their baseline HR. If a participant on the AE group is unable to progress beyond 15 min at 60%–70% hour max over the initial 2 weeks, they will be screened out of the study. After the conditioning exercise programme, subjects will continue with the intervention part of the protocol. Participants will complete AE or nAE three times a week on non-consecutive days over 4 weeks.
Aerobic exercise (AE): Participants will undergo moderate intensity AE on a treadmill over 30 min (American Heart Association recommendation for adults). HR will be monitored throughout the entire procedure by a sensor. The investigator will sequentially increase the treadmill speed by 0.1 mph every 5 s, until the participant reaches 60%–70% of age-predicted maximal heart rate (HRmax), following the formula HRmax=208 − (0.7 * age), as this has been found safe in various conditions.22 43–47 AE intensity will be modulated based on the participant’s HRmax throughout the session. If the HRmax exceeds 70%, the investigator will decrease treadmill speed by 0.1 mph every 5 s until returning to the 60%–70% HRmax target. If HRmax reaches 80% or the subject shows any signs of discomfort, the session will be stopped.
Non-aerobic exercise (nAE): Participants will walk on the treadmill for 30 min with a workload intensity within 5% baseline HR, as we used this method in our preliminary study.37
As recommended by ACSM guidelines for AE in patients with FM, the participant will be questioned regarding any respiratory or cardiovascular symptoms on each visit before starting the exercise; we will monitor pain and fatigue levels after the first 5, 15 and 25 min of exercise using a numeric pain scale.48 Additionally, to evaluate adverse effects during AE or nAE training, we will record any musculoskeletal symptoms such as pain, muscle strain, muscle soreness, fatigue, dizziness and shortness of breath.
Transcranial direct current stimulation (tDCS)
A 1×1 low-intensity DC stimulator, the Soterix Medical 1×1 tDCS-Clinical Trial, will be used with codes corresponding to active or sham stimulation, allowing a double-blinded procedure. Participants will receive 16 tDCS sessions over 4 weeks of treatment. Weeks 1 and 2 will begin with five consecutive days of tDCS followed by weeks 3 and 4 with three alternating days of tDCS. The exercise and the tDCS will be performed simultaneously as explained in figure 2.
{kind=link}
{kind=link}
Schematic view of the timeline. tDCS, transcranial direct current stimulation.
Active (anodal) tDCS: During active tDCS, a 2 mA constant current will be delivered for 20 min through rubber electrodes encased in 35 cm2 saline-soaked sponges. The anode will be placed over the left primary motor cortex (M1) and the cathode over the contralateral supraorbital area. M1 will be localised using the 10/20 International EEG System (C3—adapted by measuring 5 cm below the vertex), a reliable method for tDCS.23
Sham tDCS: We will use the same montage and parameters as active tDCS, but the active current will be applied for 30 s in the beginning and at the end of the procedure to simulate the same sensations of the current ramping as in active stimulation.49 Using 30 s of ramping is reliable for blinding50 and less than 3 min of tDCS induces no cortical excitability effects.49
A tDCS adverse events questionnaire will be administered after each stimulation session. Subjects will be instructed not to use other methods of electrical stimulation during the trial.
Outcomes
Evaluation of endogenous pain inhibition system (primary outcomes)
During the CPM and TSPS protocols, heat pulses will be generated by a TSA-II Stimulator (Medoc Advanced Medical Systems, Ramat Yishai, Israel) delivered to the right proximal volar forearm using a 30 mm × 30 mm embedded heat pain (HP) thermode. A minimum interval of 10 min between the two assessments will be respected.
Conditioned pain modulation (CPM) evaluates the ability to inhibit pain. When a pain test stimulus is given together with a conditioning pain stimulus, the test stimulus is perceived as less painful than when it was given alone.51 We will follow the adapted protocol suggested by Granot et al (2008)52 and Nir et al (2011).53 We will first determine the pain-60 test temperature (which is the temperature that induces pain sensation at a magnitude of 60 on a 60–100 numerical pain scale (NPS)) by applying a Peltier thermode (Medoc Advanced Medical Systems, Ramat Yishai, Israel) on the right forearm and delivering three short heat stimuli (43°C, 44°C and 45°C), each lasting 7 s (starting from the time the stimulus intensity reaches the destination temperature). Subjects will be asked to rate the level of pain intensity using a NPS ranging from 0=‘no pain’ to 100=‘the worst pain imaginable’. If the first temperature of 43°C is considered too painful (>60/100), we will stop the series and will provide additional stimuli at lower temperatures of 41°C and 42°C. If the three temperatures (43°C, 44°C and 45°C) are unable to achieve pain-60, we will deliver additional stimuli at 46°C, 47°C and 48°C until reaching the desired pain level of 60/100; in the unlikely event that none of those temperatures elicits pain-60, we will consider it to be 48°C. On determining the pain-60 temperature, we will administer the test stimulus at that temperature for 30 s, and subjects will be asked to rate their pain intensity at 10, 20 and 30 s after the thermode reaches the pain-60 temperature (mean scores of the three pain ratings will be calculated). Five minutes after delivering the test stimulus, the conditioning stimulus will be applied: the subject’s left hand will be immersed for 30 s in a water bath set at 10°C–12°C. Then, the same pain-60 temperature will be applied to the right forearm (left hand will still be immersed) for 30 s and the subject will again be asked to rate their pain intensity three times after the thermode reaches the pain-60 temperature: at 10, 20 and 30 s (mean scores of the three pain ratings will be calculated). CPM response will be calculated as the difference between the average of pain ratings from the test stimulus minus the average of pain ratings during the conditioned stimulus.
Temporal slow pain summation (TSPS) represents summation of C fibre mediated pain, assesses central sensitivity and is used to probe pain processing abnormalities in several chronic pain disorders.54 55 Subjects will be trained to identify pain-60 test temperature (see CPM protocol above) and we will follow the adapted protocol suggested by Staud et al (2014)56 in which the HP-thermode was programmed to deliver pulses with rise/fall of 1–2 s, depending on subject’s pain-60 level, from adapting temperatures to peak temperatures, with a plateau of 0.7 s. They will receive 1 train of 15 repetitive heat stimuli at 0.4 Hz, which (being suitable to elicit TSPS in most subjects) allows the rating of individual pain stimuli and is unlikely to induce peripheral sensitisation.57 TSPS will be calculated as the difference between HP rating after the 1st and 15th stimuli.
Evaluation of cortical markers of inhibitory control (secondary neurophysiological outcomes)
Transcranial magnetic stimulation (TMS)
To assess tDCS and AE effects, we will measure the excitability of pain-related pathways using TMS markers. TMS assessments will be similar to our previous study.58 Single pulse TMS will be performed to acquire resting motor threshold (rMT) and motor evoked potentials (MEPs); paired pulse technique will measure short interval cortical inhibition (SICI) and intracortical facilitation (ICF). We will use Magstim Rapid2 device with a figure-of-eight magnetic stimulator coil placed on the right and left M1 (for all assessments) and will record surface electromyogram from the contralateral first dorsal interosseous muscle. TMS data will be recorded and stored in a computer for off-line analysis.
Resting motor threshold (rMT): Initially, we will investigate rMT following the technique described by Rossini and colleagues, where rMT is defined as the lowest stimulus intensity to evoke a MEP of 100 μV in 3/5 trials in the relaxed muscle.59
Motor evoked potential: We will initially adjust TMS machine output intensity to achieve a baseline MEP of 1 mV peak-to-peak amplitude before the intervention. Stimulation intensity will be kept constant for each subject throughout the evaluation sessions. We will record 10 MEPs for each assessment and average their peak-to-peak amplitudes and areas-under the-curve.
Short interval intracortical inhibition (SICI) and intracortical facilitation (ICF): We will use paired pulse testing with a subthreshold conditioning stimulus (80% rMT) followed by a suprathreshold test stimulus of 120% of the motor threshold. Interstimulus intervals will be 2 ms for SICI and 10 ms for ICF. Ten randomised stimuli will be applied at each interval and the percentage of inhibition or facilitation for each interstimulus interval before and after treatment will be calculated. The paired pulse MEP intensity will be the machine output intensity eliciting 1 mV peak-to-peak amplitude that day—not the baseline MEP intensity used for single pulse testing. If we cannot obtain rMT, we will not perform MEPs or paired pulse.
Electroencephalography (EEG)
EEG will take place over approximately 45 min: 25 min of participant and software preparation, 10 min of EEG recording divided into a resting EEG condition (5 min with eyes open, 5 min with eyes closed) and a task-related condition (8 min). Participants will be asked to relax in the resting condition; the investigator will ensure they do not fall asleep.
The task-related condition will include movement observation (MO), movement imagery (MI) and movement execution (ME). This will be recorded by connecting the Net Station software (for EGI) with E-Prime. The entire task-related condition part will consist of 60 trials, with 20 trials for each of MO, MI and ME in a randomised order.60 61 Each trial will involve initial fixation (on a cross on a screen), followed by a visual cue stating the task to be performed (‘imagine’ and ‘clench’), and a video will automatically play for observation. During each MO trial, the participant will view a video of a right hand clenching; during the MI task, the participant will be asked to imagine clenching her/his right hand once, and during the ME task, the subject will be asked to clench her/his right hand once. There will be a 4 s rest period between each trial. The purpose of the task-related condition is to evaluate ERD that reflects the motor cortex activation.62
We will record the EEG in a standardised way63 using the 64-channel EGI system (EGI, Eugene, USA). The EEG will be recorded with a band-pass filter of 0.3–200 Hz and digitised at the sampling rate of 250 Hz64 by connecting the Net Station software (for EGI) with E-Prime. On acquiring the EEG data, the EEGs will be inspected and artefacts will be cleaned manually. We will use EEGLAB and analysis of EEG data will include a power analysis of the power bands in the resting EEG portion—delta (1–4 Hz), theta (4–8 Hz), alpha (8–13 Hz) and beta (13–30 Hz) bands—fast Fourier transformation, Independent Component Analysis (ICA) decomposition, ERD responses of the three different motor tasks, functional connectivity measures and topographical analysis. The analysis will compare groups at baseline, during the stimulation period, on the last day of the intervention and at the 3 months follow-up.
Secondary clinical outcomes
The following secondary outcomes will be assessed: average pain intensity as assessed by Modified Brief Pain Inventory; Revised Fibromyalgia Impact Questionnaire; quality of life assessed by Quality of Life Scale, Patient Reported Outcomes Measurement Information System; Pittsburgh Sleep Quality Index and Beck Depression Inventory.
Timeline
This trial has 25 visits divided into four components (consent and pretraining walking, conditioning exercise programme, intervention and follow-up). To increase adherence to protocol, we will adjust the calendar of sessions according to the subject’s availability (figure 2).
Study sample
Our target population is individuals with FM according to the ACR 2010 criteria. We plan to enrol 148 subjects divided into 4 groups (n=37/group).
Sample size calculation
We used the information from trials measuring the effects of tDCS and AE on CPM and TSPS according to different scenarios to do this sample size calculation (table 1).
Effect size in three scenarios
In Scenario I, we considered the effects of tDCS on CPM in patients with chronic pain: this resulted in an effect size (ES) of 0.79.
In Scenario II, we evaluated the effect of tDCS on CPM in healthy volunteers: this resulted in a pooled ES of 1.02.
In Scenario III, we evaluated the effect of exercise on CPM in chronic pain and this resulted in an ES of 0.78.
Based on this analysis, we decided on a conservative approach and chose the lowest ES; thus, we used an ES of 0.78. In addition, it is important to underscore that we expect that the combination of tDCS+AE will have a higher effect than each intervention alone (tDCS, exercise or placebo). Additionally, in this current proposal, the dosage of tDCS is higher than the studies we used to calculate the sample size (see tables 1 and 2).
Two-tailed analyses
We assumed a type I error of 5% (alpha) and made a sensitivity analysis with a type 2 error (beta) of 10%, 15% and 20% (therefore a power of 90%, 85% and 80%). We used a t-test for two independent means and considered dropout rates of 20% and 15% (table 2).
Although most studies used a power of 80% and a dropout rate of 10%–15%,22 29 65–70 we chose a dropout rate of 20% and power of 85% as to be more conservative and also account for unexpected factors.
Data analysis
All data collected will be kept in a secured and password protected database, accessible only to IRB trained and approved study staff. All analyses will be performed as intention-to-treat in which all randomised subjects who receive at least one intervention session will be included. We will conduct sensitivity analyses and test different models of handling missing data: Last Observation Carried Forward and Multiple Imputation. The change in the primary efficacy endpoints, CPM and TSPS, from baseline to week 4, will be tested with a mixed linear regression model. This model will be adjusted for important demographic variables (eg, gender) and baseline clinical parameters where appropriate. All tests will be two-sided (alpha level 0.05).
We will initially test our main hypothesis that active tDCS+AE increases CPM and decreases TSPS more than sham tDCS+nAE. If the effect is significant, we will then test differences between the active tDCS+AE group versus the two interventions alone. We will run a secondary mixed linear model to estimate the rate of change over time (using the secondary endpoints added in this model—Week 2 and follow-up) and also include the interaction term (treatment*time) to detect whether treatment effect changes differently over time. If the interaction is not significant, we will then test whether there is a main effect of time that is independent of treatment level (interaction will be removed from the model). We will adjust this model for important covariates such as age, gender, pain levels (NPS) and other baseline clinical outcomes where appropriate. For secondary clinical variables with significant effects, we will test whether they moderate the interventions’ effects on our mechanistic (TMS and EEG) outcomes, thereby gaining additional mechanistic insights. To complete our analysis, we will apply a path analysis71 to CPM and TSPS to determine if endogenous pain modulation changes (indexed by CPM and TSPS) associated with active tDCS+AE is related to direct effects versus indirect effects through secondary outcome improvements. We propose that a direct effect of active tDCS and AE on the endogenous pain inhibitory system can be inferred if the treatment effect cannot be explained by changes in psychological or functional outcomes.
An independent monitoring committee will review data on recruitment, adherence and safety; meetings will occur annually, after enrolment of 25% of the target sample or in case of reports of any serious adverse events. NIH will also perform annual site monitoring visits.
Patient and public involvement
Patients and public were not involved in the design of this study.
Ethics and dissemination
This protocol was approved by the IRB at the Partners Human Research Committee (Protocol approval number: 2017P002524). All requirements regarding the welfare, rights and privacy of human subjects protection were fulfilled. The risks of this clinical trials were considered to be minimal and are addressed in the protocol and consent form. Informed consent will be obtained from all participants before any study procedures by the Principal Investigator or coinvestigators. Trial registration number: NCT03371225. For a complete list of trial registration dataset and protocol version history, please refer to online supplementary file 1.
Supplemental material
The study findings will be reported in conferences and in peer-reviewed journal publications.
References
Footnotes
LC-B and EUK are joint first authors.
Contributors LC-B and EUK are joint first authors and with FF conceptualised the paper. PG-M, LC-S and AE-G wrote the abstract. LC-B, EUK, LC-S, ME-G wrote the introduction. AC-R, CBP, IM-T, EUK, LC-B, MME, KP-B, PG-M, DD, AE-G and YY wrote methods and analysis. AC-R, IM-T and YY prepared the figures. FF, MME, WC and HR provided critical review.
Funding This work is supported by NIH grant R01 AT009491-01A1.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study obeys the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of Partners Healthcare under the protocol number 2017P002524.
Provenance and peer review Not commissioned; externally peer reviewed.