Article Text

Abstract
Introduction Delirium is a common complication of critical illness, associated with negative patient outcomes. Preventive or therapeutic interventions are mostly ineffective. Although relaxation-inducing approaches may benefit critically ill patients, no well-designed studies target delirium prevention as a primary outcome. The objective of this study is to assess feasibility and treatment effect estimates of a multimodal integrative intervention incorporating relaxation, guided imagery and moderate pressure touch massage for prevention of critical illness delirium and for related outcomes.
Methods and analysis Randomised, controlled, single-blinded trial with two parallel groups (1:1 allocation: intervention and standard care) and stratified randomisation (age (18–64 years and ≥65 years) and presence of trauma) with blocking, involving 104 patients with Intensive Care Delirium Screening Checklist (ICDSC): 0–3 recruited from two academic intensive care units (ICUs). Intervention group participants receive the intervention in addition to standard care for up to five consecutive days (or until transfer/discharge); control group participants receive standard care and a sham intervention. We will assess predefined feasibility outcomes, that is, recruitment rates and protocol adherence. The primary clinical outcome is incidence of delirium (ICDSC ≥4). Secondary outcomes include pain scores, inflammatory biomarkers, heart rate variability, stress and quality of life (6 weeks and 4 months) post-ICU discharge. Feasibility measures will be analysed descriptively, and outcomes will be analysed longitudinally. Estimates of effects will be calculated.
Ethics and dissemination The study has received approval from the Human Research Ethics Board, University of Alberta. Results will inform the design of a future multicentre trial.
Trial registration number NCT02905812; Pre-results.
- critical illness
- music therapy
- relaxation
- guided imagery
- autonomic nervous system
- delirium
- complex intervention
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- critical illness
- music therapy
- relaxation
- guided imagery
- autonomic nervous system
- delirium
- complex intervention
Strengths and limitations of this study
We will test feasibility and measures of effect of a previously piloted relaxation-inducing intervention for the prevention of delirium and improvement of related outcomes in critically ill patients.
We will employ an evidence-based, non-pharmacological multimodal integrative intervention that has shown effectiveness for reducing pain and improving a number of secondary outcomes in a previous pilot study.
This pilot aims to assess estimates of effect and feasibility to inform a future trial.
Although clinicians and outcome assessors will be blinded, due to the nature of the intervention, participants and nurses providing direct care to patients cannot be blinded to allocation, although they will be blinded to the study hypotheses.
The mechanisms of effects of relaxation-inducing interventions in critical illness are not well understood; hence, we aim to explore effects of the intervention on parasympathetic system activation and inflammatory markers.
Introduction
Intensive care unit (ICU) delirium affects 35%–55% of critically ill patients and is independently associated with a 13-fold (adjusted OR: 4.88–13.0) increased risk of death1 and long-term cognitive impairment.2 ICU delirium carries important financial and societal burdens ((39% higher adjusted ICU (95% CI 12% to 72%) and 31% higher hospital costs (95% CI 1% to 70%)).3 Moreover, patients identify frightening delirium experiences and pain as the most severe stressors4 5 in critical care. Pharmacological interventions for the prevention and treatment of delirium have limited benefit and are associated with high costs and risks for side effects.6 7 Although clinical guidelines recommend the development of non-pharmacological interventions to prevent delirium,8 effective prevention strategies have yet to be established.9
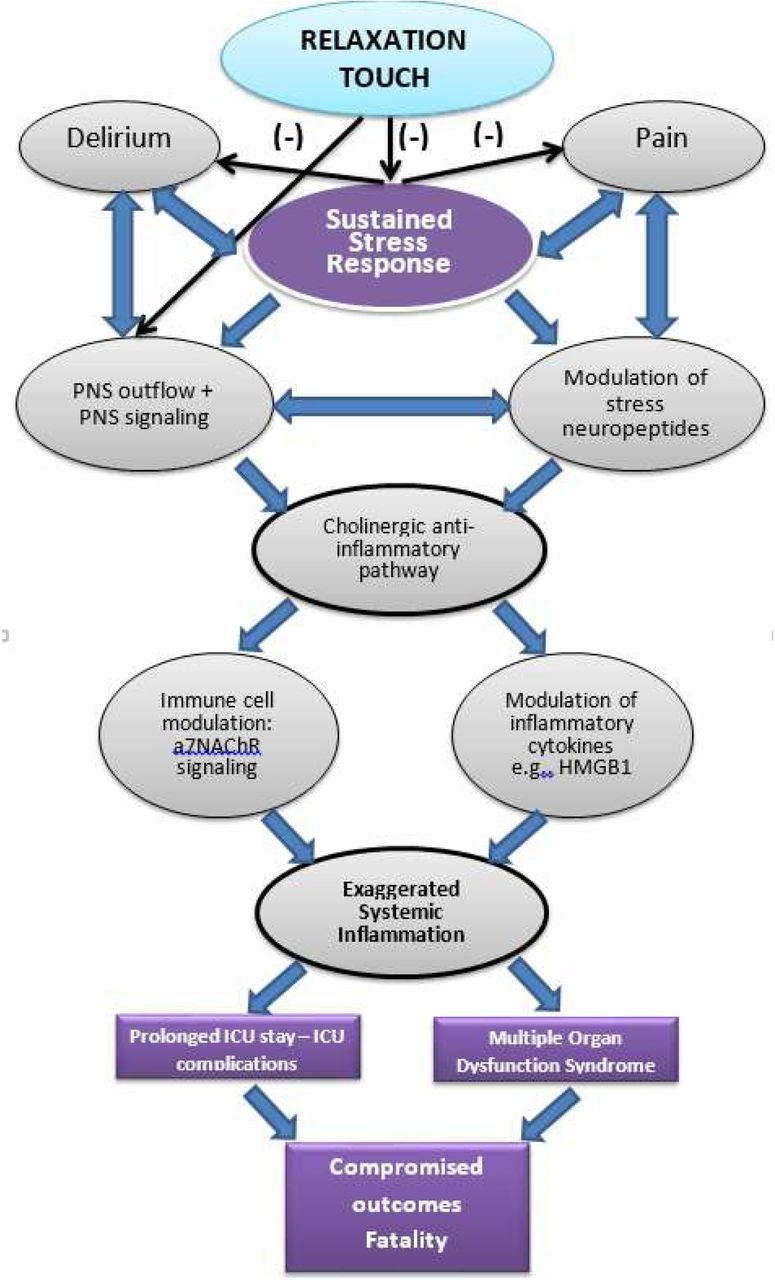
A prolonged and eventually aberrant stress response and depressed parasympathetic (parasympathetic nervous system (PNS)) activity have been postulated as the pathophysiological basis for the development of both ICU delirium and systemic inflammation.10–13 Frightening hallucinations and ideations during delirium may further exaggerate the stress response and prolong critical illness with detrimental consequences. Pain may worsen matters by reciprocal incremental feedback on inflammation and stress.4 14 Thus, in critical illness, stress, delirium, pain and systemic inflammation may comprise a self-perpetuating syndrome. Attenuating the cascade of negative health impacts from pain and delirium has become a high clinical priority (figure 1).8 Moreover, the growing recognition that delirium and other critical illness sequelae may have long-term consequences in critical illness survivors15 further highlights the need for prevention strategies.

Evidence-based framework for the physiological mechanism implicated in relaxation-induced effects in critical illness. Relaxation acts early at the pathophysiological cascade through which an exaggerated stress response results in proinflammatory effects, suppressed PNS outflow and subsequently in systemic inflammation, multiple organ dysfunction and death. The relaxation response counterbalances the exaggerated stress response and activates PNS and cholinergic anti-inflammatory signalling, which downregulates proinflammatory (eg, HMGB-1) and upregulates anti-inflammatory cytokines, therefore attenuating systemic inflammation and its detrimental organ effects. α7-nAChR, alpha7-nicotinic acetylcholine receptor; HMGB-1, high mobility group box 1; ICU, intensive care unit; PNS, parasympathetic nervous system.
Evidence-based theoretical work postulates that the multitude of psychological stressors in critically ill individuals may contribute to the development of pathophysiological sequelae.10 Moreover, animal models illustrate that PNS stimulation and acetylcholine (ACh) release suppress inflammation and decrease fatality, via the cholinergic anti-inflammatory pathway.16 Devising ways to draw on the autonomic nervous systems’ (ANS) inflammation and stress regulatory properties by non-pharmacological interventions has, in theory, the potential to improve outcomes with low side effect risk. However, stimulation of the PNS in critical care is challenging. Relaxation-inducing interventions can induce PNS activity. Such approaches have successfully been used in diverse patient populations to counter stress but remain under-tested in critical illness.17 A recent systematic review shows favourable effects of relaxation and guided imagery (RGI) intervention in reducing pain, anxiety and length of stay in critically ill patients,18 whereas the relaxation-inducing effects of music in critical care have been well supported by evidence.19 In a pilot randomised controlled trial of the effects of a similar multimodal intervention on the incidence of pain and on a number of secondary outcomes, we observed significant decreases in pain incidence (RR=0.56, p=0.003) and severity (p<0.0001), systolic arterial pressure, anxiety, along with improved sleep quality.20
Hypotheses
Overall, we hypothesise that a multimodal intervention incorporating RGI and moderate-pressure touch massage is feasible within a critical care setting and can have an effect on decreasing delirium and in improving physiological and psychological outcomes in randomised critically patients who will receive standard care and the intervention compared with patients receiving standard care plus a sham intervention only.
Primary hypotheses
We hypothesise that the intervention will be feasible and that it will have an effect on decreasing delirium incidence and duration.
Secondary hypotheses
We hypothesise that the intervention will have an effect on: (A) incidence rate of subsyndromal delirium, time to delirium occurrence and proportion of delirium-free time during up to 8 days of ICU stay; (B) sedation levels and daily sedative, analgesic and antipsychotic agent dose; (C) pain occurrence and intensity; (D) perceived stress level; (E) sleep duration and quality; (F) anxiety; (G) length of ICU stay and duration of mechanical ventilation/proportion of mechanical ventilation-free days; (H) hospital length of stay (LOS), (I) physiological biomarkers; and (K) quality of life after ICU discharge.
Design and methods
Study design
Relaxation for Critically ill Patient Outcomes and Stress-coping Enhancement is a pilot feasibility, randomised, controlled, single-blinded trial with two parallel groups (intervention and standard care). Accounting for major risk factors of delirium,15 stratified randomisation according to age (18–64 years and ≥65 years) and presence of either surgical or trauma injury with blocking and 1:1 allocation to assure balance in numbers per group will be employed.
Research objectives
Research objectives include to:
Assess clinical trial feasibility with predefined goals (enrolment, randomisation, adherence, timing of intervention and workload).
Calculate estimates and variance of treatment effect across outcome measures.
Calculate CI of incidence proportions, means and SD of outcome measures in study groups.
Explore the feasibility of identifying underlying physiological mechanisms.
Setting, recruitment and sample size
Consecutive patients admitted to two academic ICUs, in Edmonton, Alberta, Canada, with an Intensive Care Delirium Screening Checklist (ICDSC) score of 0–3 will be screened for study eligibility and will be recruited by research staff at each site. In cases where an ICDSC score cannot be obtained on admission, we will screen patients for up to 4 days after admission. Since delirium occurs most often within the first 5 days of admission, screening and enrolment will take place as soon as possible and within 96 hours after ICU admission. This pilot is not powered to determine a difference in a primary outcome, since we aim to assess estimates of effect. For a definitive trial, we would require 290 (145/arm) patients to detect a 10% difference in incidence proportion between intervention (20%) and control (30%) arm (two-sided alpha=0.05, power=80%, dropout rate=10%). The Pan method,21 which is based on generalised estimating equations, was used to perform the sample size calculation under the assumption of AR1 correlation structure among 5 days repeated measurements with correlation between any two adjacent observations from the same subject of 0.5. Since this is a pilot, aiming to explore feasibility and estimates of effects, and the incidence rates used for the calculation might not be appropriate, we used 36% of the sample size of the full study to estimate the parameters accurately and get an experience for a full trial.22
Eligibility
Inclusion criteria: (A) age over 18 years, (B) ICDSC: 0–3 and (C) written informed consent by participant or by family member/surrogate in case participant not capable.
Exclusion criteria: patients: (A) already in the ICU for more than 96 hours, (B) with ICDSC >3 within 72 hours of screening in case intervention has not been initiated, (C) on special contact precautions (ie, Methicillin-resistant Staphylococcus aureus (MRSA), Vancomycin-resistant enterococci (VRE) and HIV), (D) with expected ICU LOS <72 hours, (E) with acute neurological illness/neurological trauma, persistent deep sedation or coma (Richmond Agitation Sedation Scale (RASS=−4 and –5)), (F) with current history of severe mental health problems and dementia, as per history, (G) with hearing impairment or conditions not permitting use of headphones, (H) on neuromuscular blockers, (I) with known or suspected substance/alcohol withdrawal and (J) enrolled in trials of sedatives and antipsychotics.
Patient and public involvement
The protocol is based on a pilot study with 60 randomised patients,20 12 of which provided feedback regarding the desirability, burden and specific components of the intervention, study procedures and preferred outcomes. Participants’ feedback informed the design of the study, resulting in many significant changes. Preferred post-ICU follow-up times were informed by an informal advisory group through the Alberta Innovates’ Strategy for Patient Oriented Research network of patient representatives. Moreover, as a patient and clinician engagement strategy, an advisory group with representatives of patients, families and clinicians will act as a consultation group for the research team. Acceptance and desirability of the protocol by clinicians and patients/families will be assessed further by focus group discussions. Recommendations will inform future development of the intervention and research design. Canadian Institutes of Health Research guidelines for patient engagement will be used to establish and facilitate the advisory group. Feedback on: (A) concerns on the intervention, study processes and contamination of the control group by implementation of aspects of the intervention, (B) desirability of intervention (massage, music choices, complexity, voice of recording and pace), (C) timing within the day, (D) duration/feasibility of the intervention, (E) burden of study processes, (F) outcomes to be addressed in the future trial, (G) interpretation of results and (H) strategies for dissemination of findings will be collected.
Intervention
The choice of a multimodal intervention (duration: 55 min) was based on an evidence-based literature review, its superiority to unidimensional approaches,23 the recommendations of the American Holistic Nurses Association24 and a successful pilot.20 According to the UK Medical Research Council guidance for complex intervention trials,25 development of the protocol included extensive theoretical work,10 two systematic reviews,18 26 modelling of outcomes, extensive consultations with groups of experts, one small feasibility and acceptability pilot (n=10 participants) and one larger pilot (n=60)20 and consultation with pilot participants and patients’ representatives. The intervention has been developed by the research team and a group of experts based at the University of Alberta and Cyprus University of Technology. It includes: (A) a brief moderate-pressure massage session (massage: 15 min) and (b) RGI (30 min, through headphones). The 30 min recorded RGI intervention involves: (A) guided relaxation, (B) a structured guided imagery script supported by instrumental music and (C) recorded instrumental music for 15 min (Haydn concerto no 1 in C major and Bach violin concerto in D minor, 60 beats per min approximately). The same recorded RGI script will be used at all sessions. It includes positive suggestions and instructions for gradual relaxation, followed by guidance to visualise one’s body being healed. Moderate-pressure (4 N, approximately or patient pressure rating 3/10) low velocity (1–5 cm/s) massage consists of broad and repetitive circular movements with wide area of contact, applied sequentially for 2–3 min at each site: hands, forearms, lateral arms and then over trapezius muscles, the temple, scalp, face and forehead area. Areas are to be contacted as appropriate to each participant, and the protocol may be adapted taking into account safety issues (ie, to avoid area around intravascular catheter or injury). Moderate pressure massage is involved in PNS activation, in contrast to light pressure.27 The intervention will be administered once daily (09:00–15:00) for up to five consecutive days by trained research staff not involved in patient care, who will be randomly audited by the Trial Steering Committee (TSC) to ensure protocol adherence. The decision to deliver the intervention for the first 5 days after enrolment only was based on epidemiological data regarding the onset of delirium,9 data on median length of stay of the target populations in the study institutions and cost considerations. However, it will be important to study the effect of the intervention on longer stay patients in future trials. The decision to deliver only one intervention daily was based on a small acceptability/feasibility pilot and pragmatic considerations for knowledge translation and future implementation of this approach, as well as cost considerations and burden to participants and units. The durations of guided imagery, music listening and touch intervention were based on the acceptability pilot, expert opinions on the minimum duration to achieve a relaxation effect and on published evidence.18 Despite evidence on the importance of providing choice to patients regarding music listening,19 we will use standardised music for intervention stability purposes. It will be important to look at the effect of patient-directed music in future trials of this intervention.
Interruptions and deviations from the intervention protocol will be recorded in detail. The intervention will be terminated on a patient’s transfer or discharge from the ICU. The intervention may be discontinued in case of adverse events related to the intervention or withdrawal of consent. If we observe ICDSC scores above 3 once the intervention has been commenced, the intervention will be continued, unless otherwise indicated by a participant’s condition. Although contamination of the control group by spontaneously mimicking aspects of the intervention by healthcare personnel cannot be excluded, healthcare providers in the units are very familiar with clinical trials and have been instructed on the need to abstain from mimicking the intervention. Additionally, the touch component of the intervention requires specific training and it is unlikely to be successfully replicated.
Randomisation and allocation concealment
Participants will be randomly assigned to either control or intervention group (1:1 allocation) as per a computer-generated randomisation schedule, generated by the EPICORE, University of Alberta, stratified by site, age (18–64 years and ≥65 years) and presence of surgery or trauma using permuted blocks of random sizes. The block size will not be disclosed to ensure concealment. After baseline measurements, allocation will be disclosed only to the intervention staff. Codes will be generated prior to the beginning of the study by EPICORE.
Blinding
Investigators, physicians and nurses (when possible), data collectors, research assistants and laboratory technicians will be blinded to group allocation during the trial and analysis. Due to the nature of the intervention, participants cannot be blinded to allocation. Also, participants’ primary nurses cannot be blinded to study procedures (ie, delivery of massage); however, they will remain blinded to study hypotheses and study design (ie, study outcomes and numbers and types of participants’ groups), to minimise performance bias. We believe that these procedures along with the sham intervention will maintain an adequate level of concealment even among participants’ primary nurses to minimise bias. The same intervention personnel will be involved in the delivery of the intervention and sham intervention. Intervention personnel are not involved in assessment of patient outcomes and have been trained on the importance of preserving blinding, therefore minimising the risk for potential bias.
Treatment arms
Patients randomly allocated to the intervention group will receive the intervention in addition to standard care (standard care+multimodal intervention group). Patients allocated to the control group will receive standard care and a sham intervention consisting of presence of a research staff at the bedside with drawn curtains and silent headphones (standard care+sham intervention group; schematic of study design in figure 2).
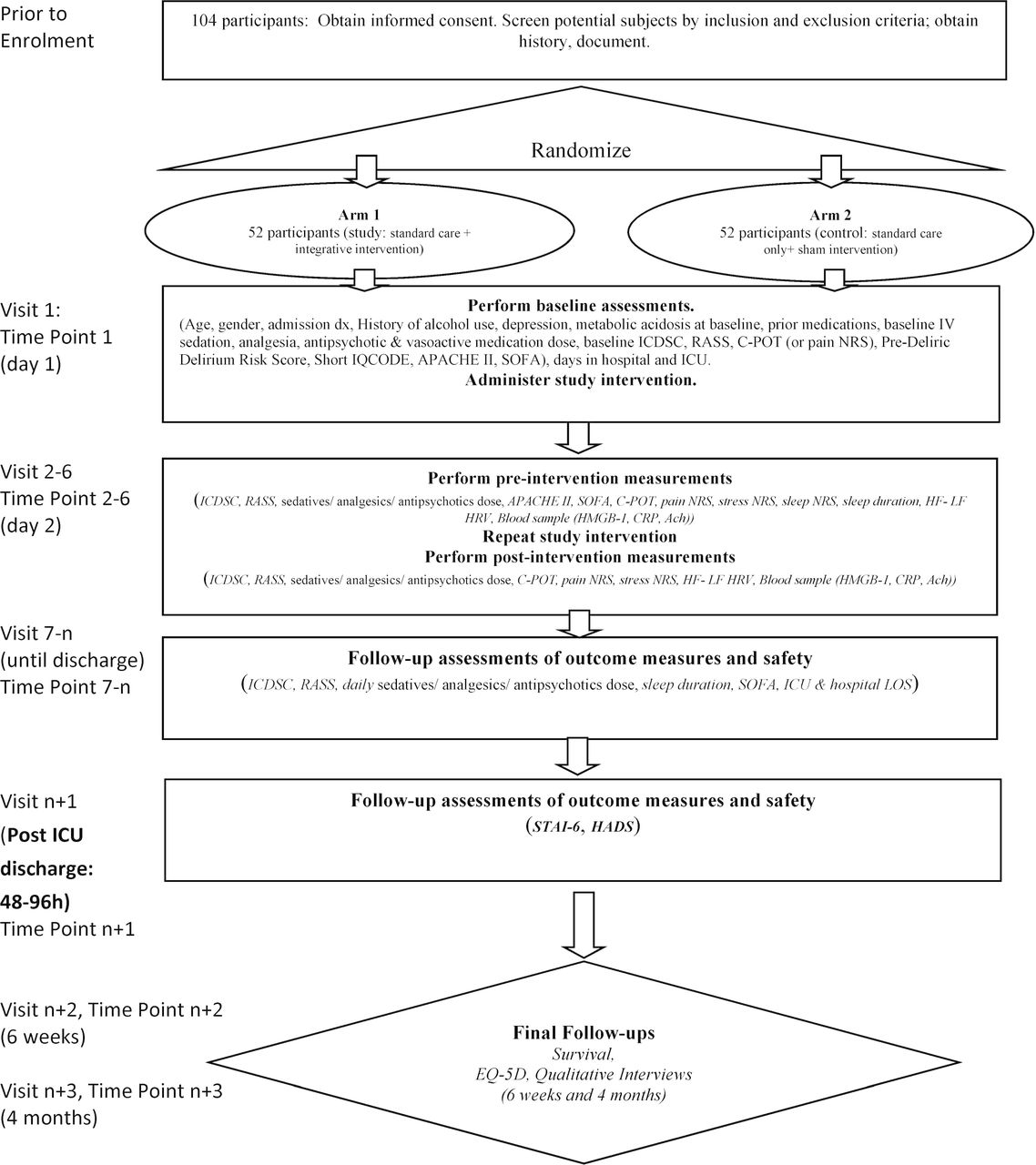
{kind=link}
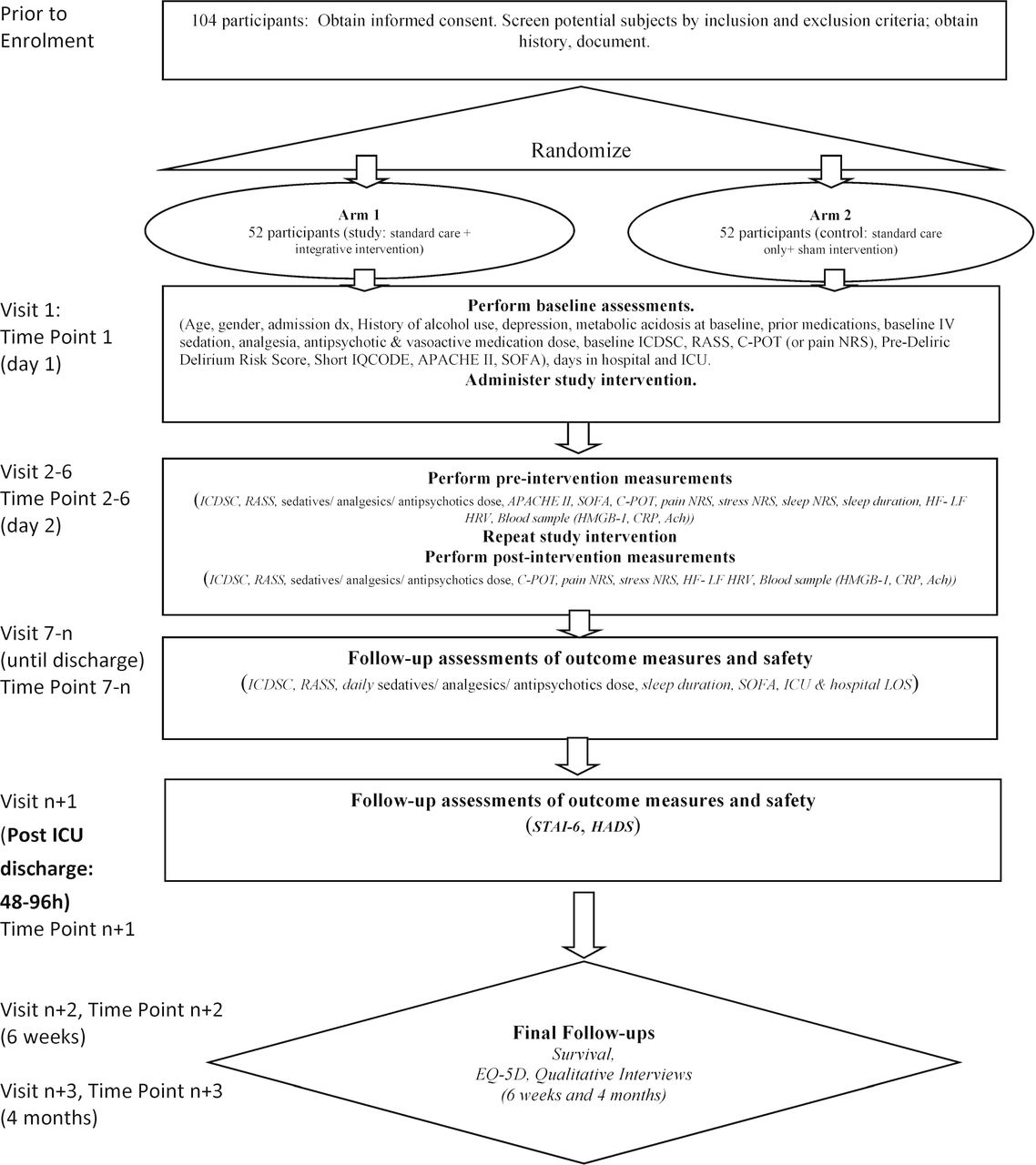{kind=link}
Schematic of study design. Ach, acetylcholine; APACHE, Acute Physiology & Chronic Health Evaluation; C-POT, Critical Care Pain Observation Tool; CRP, C reactive protein; EQ-5D, EuroQol Five Dimensions Questionnaire; HADS, Hospital Anxiety and Depression Scale; HF, high frequency; HMGB, high mobility group box; HRV, heart rate variability; ICDSC, Intensive Care Delirium Screening Checklist; ICU, intensive care unit; IQCODE, Informant Questionnaire on Cognitive Decline in the Elderly; IV, intravenous; LF, low frequency; LOS, length of stay; NRS, numeric rating scale; RASS, Richmond Agitation Sedation Scale; SOFA, sequential organ failure; STAI, State Trait Anxiety Inventory.
Concomitant care
Standard care will be continued for all participants. Type and dose of all administered sedative, psychoactive and analgesic medication will be recorded.
Duration of participation
The total duration may vary according to ICU length of stay and will be 17 weeks approximately, from enrolment until the last follow-up at 4 months post-ICU discharge.
Data collection and instruments
Data will be collected for each participant by blinded data collectors and captured in a Research Electronic Data Capture (RedCap) database developed and monitored by EPICORE. Time points include: baseline measurements, follow-up while in the ICU for up to 5 days, follow-up 48–96 hours after ICU discharge, follow-up 6 weeks and 4 months after ICU discharge. In case of participants who discontinue participation, data already collected will be retained.
All study scales are routinely used in clinical practice and have established psychometric properties. The ICDSC is one of the most reliable tools for assessment of ICU delirium advocated by recent guidelines.4 28 Interobserver reliability of and sensitivity (80.1, 95% CI 73.3 to 85.8) of ICDSC have been established and will be further assessed in this study.28 For comatose, deeply sedated patients (RASS=−4 and –5) delirium cannot be assessed. Data captured on case reports forms are included in figure 2. Baseline data captured at enrolment will include: sociodemographic data, admission diagnosis, history of alcohol use, medications prior to admission, metabolic acidosis, ICDSC, RASS, Short Form Informant Questionnaire on Cognitive Decline in the Elderly, Pre-Deliric Delirium Risk Score, baseline intravenous sedation, analgesia and antipsychotic dose, disease severity at admission (Acute Physiology & Chronic Health Evaluation II (APACHE II), Sequential Organ Failure Assessment (SOFA)), number of days in hospital and ICU prior to protocol enrolment. For pain, scores obtained at the first preintervention measurement will be considered as baseline.
Outcome measures
Primary and secondary outcome, as well as feasibility, measures include (online supplementary appendix 1):
Supplemental material
Primary clinical outcome
Incidence rate of delirium (ICDSC ≥4), during 5 days of intervention (first 5 days of enrolment). Presence of delirium will be assessed by blinded trained nurse assessors two times daily (08:00 and 20:00).
Secondary outcomes (estimates, variance of effects, proportions and means, and SD (where applicable) per group)
Delirium-related secondary outcomes: (A) incidence rate of delirium (ICDSC ≥4) during ICU stay and postintervention, (B) incidence rate of subsyndromal delirium (ICDSC: 1–3) during the intervention period and subsequent ICU stay, (C) time to delirium occurrence, (D) proportion of delirium-free time during up to 8 days of ICU stay (excluding periods with deep sedation and coma), (E) sedation levels (RASS score and (F) daily sedative (benzo-equivalents and propofol), analgesic (morphine equivalents) and antipsychotic agent (type, mg/24 hours) dose.
Pain-related outcomes (preintervention and postintervention): (A) pain intensity (self-reported (S-R) numeric rating scale (NRS) for patients able to self-report, pain indicators (Critical Care Pain Observation Tool in patients unable to self-report and (B) perceived stress level (S-R NRS).
Sleep (daily): (A) sleep quality (S-R NRS) and (B) sleep duration (in min) ((sleep monitors (Fitbit Alta HR: daily sleep cycle) and nurses’ log (duration of sleep between 19:00–07:00)). Although more reliable, due to considerations around burden to the unit and cost, we do not use polysomnography in this pilot.
Disease severity (daily): SOFA score.
Physiological biomarkers (preintervention and postintervention): (A) serum inflammation biomarkers (high mobility group box 1 (HMGB-1) and C reactive protein (CRP) levels), (B) high frequency (HF) and low frequency (LF) components of heart rate variability (HRV) as measures of PNS status and (C) serum ACh levels, as a measure of PNS activation. The choice of biomarkers was based on the theoretical framework guiding this work (triggering of a relaxation parasympathetic response and attenuation of the inflammatory response; figure 1) and results of the pilot study,20 29 which showed alterations in biomarkers immediately postintervention and overtime, despite great variability in biomarker levels. The results will help us generate more informed hypotheses for a subsequent trial.
Psychological outcomes (2–7 days after ICU discharge): anxiety (Hospital Anxiety and Depression Scale and State Trait Anxiety Inventory-6):
Clinical outcomes (at discharge): (A) length of ICU stay (ICU LOS) (or ward-ready), (B) duration of mechanical ventilation/proportion of mechanical ventilation-free days, (C) survival, (D) hospital LOS and (E) (3 months post-ICU discharge): 90-day survival.
Quality of life outcomes (6 weeks and 4 months post-ICU discharge): EuroQol Five Dimensions Questionnaire and Short-Form 36 Health Survey.
Recollection and perception of the intervention (2–7 days after ICU discharge and 6 weeks post-ICU discharge): qualitative open-ended questions to explore recollection of intervention, views on intervention and acceptability (online supplementary appendix 2).
Supplemental material
Feasibility
Acceptability: number of patients refusing or wishing to discontinue a session. Acceptability will be further assessed with the patients and clinicians advisory group.
Enrolment and consent: (A) percentage (%) of eligible patients and reasons for non-eligibility, (B) time from admission to enrolment, (C) recruitment rates and (D) percentage of patients declining consent.
Randomisation and concealment: (A) time from enrolment to randomisation, (B) percentage of of cases at which allocation was inappropriately revealed and description of incident.
Protocol adherence and intervention fidelity: percentage of: (a) participants completing the entire study protocol, (B) sessions missed, interrupted and delayed, and reasons. Adherence to intervention protocol assessed by random observation audits and by intervener’s detailed reports of any deviations from protocol and related reasons.
Data collection and management: (A) timeliness, accuracy of data collection and reliability, (B) testing of trial database, (C) type and percentage of missing values, (D) qualitative data on participants’ perceptions of the study and (E) time required for all study procedures and intervention (to be used in future economic assessment and assessment of burden of the trial).
Primary feasibility criteria
The study will be considered feasible for the specific population and ICU context if the following five criteria are met:
Eligible patients declining consent <60%.
Cases at which allocation was inappropriately revealed <3%.
Participants withdrawing from study protocol while still in the ICU <10%.
Average sessions missed per patient <40%.
Average sessions interrupted per patient <50%.
Biomarker quantifications
Blood sampling
One 5 mL blood sample will be collected in precoded general anticoagulated phials, through an intravascular catheter already in place, within 10 min before and 10 min after the intervention.
Biomarker analyses
Serum levels of HMGB-1, ACh and CRP
These will be quantified by commercially available sandwich ELISA kits (LifeSpan Biosciences, Seattle, Washington, USA; Biosource, S. Diego, California, USA; R & D Systems, Minneapolis, Minnesota, USA< respectively). Intra-assay and interassay coefficients of variation are expected to be less than 10%. All samples will be tested within the same assay run in duplicate by a specialised laboratory technician. Measurements will be carried out at the Women’s Health Research Laboratory, Faculty of Nursing, University of Alberta.
ANS activity: HF, LF AND LF/HF ratio
ANS activity will be assessed through frequency domain analysis of ECG recordings. ECG recordings will be logged using the Zephyr BioModule, and data will be imported into the OmniSense software. Data from OmniSense will then be uploaded to Kubios HRV software (Kubios HRV – Hear Rate Variability Analysis Software), and HF, LF and LF/HF ratios will be computed. HRV data will be recorded continuously starting from 5 min before, during and 5 min after the intervention. For ease of incorporation into statistical models, HRV components will be analysed at 5 min before the intervention, 5 min after the intervention and several times during the intervention based on the timing of its various components. Specifically, HRV components will be will be analysed on the 8th and 15th min of massage (half-way and conclusion of massage), 3rd and 15th min of RGI (induction and conclusion of RGI) and end of music therapy.
Data analysis
Statistical methods
Demographic/clinical characteristics of patients and all outcomes will be presented by treatment group using descriptive statistics: mean (SD), median (IQR) or proportion. Outcomes will be analysed longitudinally over 5 days by logistic regression model based on generalised estimating equations (GEE) with first-order autoregressive correlation structure (AR1) correlation structure. Analysis of covariance, t-test or Mann-Whitney U test, as appropriate, will be conducted for the continuous outcomes that are not longitudinal. Models will be adjusted for covariates including predeliric score, administration of medication (sedatives, analgesics, vasoactives and antipsychotics) and severity score (SOFA and APACHE). χ2 or exact test, as appropriate, will be used for categorical outcomes. We will treat pain outcomes as both categorical (presence or absence of pain) and continuous variables (pain score). CIs will be presented with estimated effects. Primary analysis will be based on all available data using data from all assessments. Since GEE assumes missing completely at random (MCAR) mechanism, we plan to conduct a sensitivity analysis based on inverse probability-weighted GEE,30 which employs a less restrictive missing at random (MAR) mechanism. A ‘last observation carried forward’ (LOCF) approach was not considered because analysing all available data performs better than LOCF in GEE setting with respect to bias, type I error rate and coverage probability under both MCAR and MAR mechanisms. Although we are not able to formally assess potential effects of the sham intervention with the current two-group design, GEE models will provide indications of such potential effects that will be taken into account at a future trial. Analysis will be conducted by EPICORE.
To account for non-compliance, protocol deviations and missing outcomes, intention-to-treat (ITT) analysis will be employed. ITT analysis includes every randomised subject according to treatment assignment. Additionally, per-protocol (PP) analysis will also be employed. PP population is defined as a subset of the ITT population who completed the study without any major protocol violations. If ITT and PP analyses lead to similar conclusions, the reliability of results will be supported. Although this is a pilot trial, ITT will help reduce potential effects of selection bias. PP analysis will assess whether the ITT result is too conservative. This will provide important data regarding the effect size for the subsequent trial. Both analyses will assist us in revising the protocol for the larger trial.
Qualitative analyses
Interview transcriptions will be thematically analysed by an inductive content analysis approach.31 A coding scheme will be developed based on recurrent themes of the first five interviews. Subsequently, two researchers (EDEP and TP) will code independently, using axial and inductive coding to formulate a final coding template by consensus. The final coding scheme will then be used to code, compare and interpret all transcripts. Individual analyses of the team members will be discussed to achieve shared understanding and to increase reliability. The data will be analysed via NVivo software (QSR International Doncaster, Victoria, Australia).
Quality control and quality assurance
This pilot will be supervised by an independent TSC, consisting of three clinical trial experts independent of the research team. Periodical audits of the intervention and trial processes at both sites by personnel independent from investigators will be initiated by the TSC. Randomisation, recruitment, intervention adherence, blinding, stability and data collection processes will be monitored. Trial Monitoring Committee will also review relevant information from similar studies and will consider the recommendations of the Data Monitoring and Ethics Committee. Study personnel have been trained to standardise processes. We have developed two training videos including detailed demonstration of the intervention. Moreover, research personnel involved with the intervention has received 18–21 hours of hands-on training, including detailed auditing, and several hours of self-paced training to standardise the process, communication and timing of intervention. Study personnel will meticulously record any deviations from the intervention protocol to assess feasibility and effect on outcomes. Clinical data will be retrieved from the quality-controlled clinical information system of the units. Detailed electronic data collection forms with embedded quality controls will be used and reviewed in detail. Data quality will be monitored through EPICORE before, during and after entry. All data will be entered electronically using study forms generated through RedCap with embedded quality control processes. Study data will be collected and managed using REDCap electronic data capture tools hosted at the University of Alberta.32 A quality control system will be applied for biological measurements as per lab protocol. Day-to-day operations of the trial will be overseen by a Trial Management Group (TMG) comprising, as a minimum, the principal investigator, senior trial manager, trial manager, trial statistician and data manager. TMG meetings will take place on a regular basis throughout the duration of the study. The TMG will have responsibility for ensuring the adherence and progress of the study in relation to all regulatory, administrative academic and any clinical or safety issues.
Ethics
This study will be conducted in compliance with Canadian and International Good Clinical Practice standards. No deviation from the protocol will be implemented without the prior review and approval of the Human Research Ethics Board (HREB) except where it may be necessary to eliminate an immediate hazard to a research participant. In such case, the deviation will be reported to the HREB.
Experienced research personnel not involved with the delivery of the intervention and in patient care will acquire informed consent from legal surrogates or participants if competent to consent. Participant assent will be acquired when participants regain capacity. Confidentiality, anonymity and right to withdraw at any point with no questions asked and no effect on the quality of care received will be assured. After completion of the study, the data and samples will remain stored at the academic institution for 5 years.
Confidentiality
Code-identified encrypted study data will be stored separately from participant information at an EPICORE database permitting code-access only. All study forms, lab specimens and data will be identified by an alphanumeric code to maintain confidentiality. Records that contain names and identifiers will be stored separately from study data identified by code. Participants’ information will not be released outside the study. Participant information will be stored at an encrypted limited code-initiated access electronic file. Consent forms will be kept in a locked file cabinet at a prespecified limited-access room at the University of Alberta.
Safety
Although prior pilot data did not provide evidence of adverse effects or increased rate of complications, any physiological/behavioural alteration during interventions will be recorded and analysed. Adverse events, irrespective of causal relationship, will be collected for all participants during and up to half an hour after the intervention.
Data dissemination
Results will be disseminated to participants, healthcare professionals, health services authorities and the public via conference presentations and publications. Results of this study will be used to inform the design and conduct of a future multicentre trial. The results of the trial will be presented at national and international meetings and published in peer-reviewed journals. A lay summary of the results will be available to trial participants on request. An online summary of the findings will also be made available.
Conclusions
This pilot clinical trial integrates a low-risk, patient-centred strategy, translational research and psychological outcomes to allow an evaluation of non-pharmacological delirium management with mechanistic insights. Implications of the definitive trial include the potential to reassure patients, decrease the incidence of frightening delirium experiences and improve longitudinal outcomes.
Acknowledgments
The authors are grateful to the participating sites: Royal Alexandra Hospital, Edmonton, Alberta, Canada, and University of Alberta Hospital, General Systems ICU, Edmonton, Alberta, Canada. We would like to acknowledge the support of the Consultation and Research Services Platform at The Alberta SPOR SUPPORT Unit in the statistical services. We wish to thank the Epidemiology Coordinating Research Centre (EPICORE), University of Alberta, for statistics consultation, and Lily Yushko, systems analyst at EPICORE for development of patient report forms and Thilini Herath Mudiyanselage for contributions to the final version of the manuscript.
References
Footnotes
Contributors EDEP, YS, KH and DJK contributed to study conception, design and manuscript draft. HTS, CN, LR, SMB, MM and TP contributed to discussions about design; EDEP, YS, KH, DJK, HTS, CN, LR, SMB, MM and TP are investigators in the Canadian Institutes of Health Research (CIHR) grant supporting this work. PT contributed to refining data collection strategies. CL and RA contributed to the development and teaching of the massage intervention, and IH contributed to randomisation and statistical analysis plan. TSB contributed to the development of the music therapy piece of the intervention. This study is supported by the Canadian Critical Care Trials group. All authors contributed to refinement of the study protocol and approved the final manuscript.
Funding This work is supported by a CIHR grant (CIHR PS 148850; PI: EDEP) and an Alberta Innovates Strategy for Patient Oriented Research (SPOR) Support Grant (PI: EDEP).
Competing interests None declared.
Patient consent Not required.
Ethics approval The protocol has received approval from the Human Research Ethics Board University of Alberta and administrative approvals from participating institutions.
Provenance and peer review Not commissioned; externally peer reviewed.