Article Text

Abstract
Introduction Preterm birth (PTB) results from heterogeneous influences and is a major contributor to neonatal mortality and morbidity that continues to have adverse effects on infants beyond the neonatal period. This protocol describes the procedures to determine molecular signatures predictive of PTB through high-frequency sampling during pregnancy, at delivery and the postpartum period.
Methods and analysis Four hundred first trimester pregnant women from either Myanmar or Thailand of either Karen or Burman ethnicity, with a viable, singleton pregnancy will be enrolled in this non-interventional, prospective pregnancy birth cohort study and will be followed through to the postpartum period. Fortnightly finger prick capillary blood sampling will allow the monitoring of genome-wide transcript abundance in whole blood. Collection of stool samples and vaginal swabs each trimester, at delivery and postpartum will allow monitoring of intestinal and vaginal microbial composition. In a nested case–control analysis, perturbations of transcript abundance in capillary blood as well as longitudinal changes of the gut, vaginal and oral microbiome will be compared between mothers giving birth to preterm and matched cases giving birth to term neonates. Placenta tissue of preterm and term neonates will be used to determine bacterial colonisation as well as for the establishment of coding and non-coding RNA profiles. In addition, RNA profiles of circulating, non-coding RNA in cord blood serum will be compared with those of maternal peripheral blood serum at time of delivery.
Ethics and dissemination This research protocol that aims to detect perturbations in molecular trajectories preceding adverse pregnancy outcomes was approved by the ethics committee of the Faculty of Tropical Medicine, Mahidol University in Bangkok, Thailand (Ethics Reference: TMEC 15–062), the Oxford Tropical Research Ethics Committee (Ethics Reference: OxTREC: 33–15) and the local Tak Province Community Ethics Advisory Board. The results of this cooperative project will be disseminated in multiple publications staggered over time in international peer-reviewed scientific journals.
Trial registration number NCT02797327; Pre-results.
- obstetrics
- fetal medicine
- maternal medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The prospective study design by building a pregnancy–delivery cohort.
The nature of the proposed systems approach, a holistic approach in which investigators do not need to choose the parameters that will be measured as it will by default simply be ‘everything’, and it is in that sense that these approaches are inherently unbiased, is a strength on one hand but has its limitations on the other hand (eg, gene panel selection bias or technical bias such as sequencing bias in library construction or amplification bias).
High-frequency sampling throughout the entire pregnancy, facilitated by enrolment in the first trimester, which is ensured by precise estimation of gestational age via early ultrasound scan for all participants, enables to evaluate transcript abundance in the peripheral whole blood that contains granulocytes and mononuclear cells in a genome-wide scale.
The fact that women with presumably complicated pregnancies or women who potentially require a caesarean section (eg, history of caesarean section) are excluded from the study since advanced medical services due to the rural setting and access to clinic-based but only limited access to hospital-based facilities might introduce a selection or exclusion bias.
Bias due to missing data and protocol deviations: resulting from the high-frequency sampling, it is expected that some women will not be able to provide all per-protocol samples.
Introduction
Preterm birth (PTB), defined as delivery before 37 weeks of gestation, is a major cause of neonatal mortality and morbidity.1 2 In 2010, the estimated number of neonates born before 37 weeks of gestation was 14.9 million worldwide, which translates into an incidence of 11.1% of all live births, with higher rates in developing countries.3 PTB is the most important cause of neonatal death worldwide and accounts for a large proportion of under-5 infant mortality.4 5 In addition to neonatal mortality, the proportion of preterm infants with long-term physical and mental impairments is higher when compared with term neonates.6 Not surprisingly, survival of preterm neonates is associated with the gestational age at delivery and the socioeconomic setting at birth as there is a huge gap when comparing survival in low-income and middle-income countries (LMICs) to high-income countries.3 7 The mortality risk of preterm infants compared with term infants in the proposed study population is significantly higher for early premature infants defined as delivery at ≥28–<34 weeks of gestation (OR 9.5, 95% CI 5.4 to 16.5, p<0.001), while the mortality risk for late premature infants defined as delivery at 34 to <37 weeks of gestation (OR 1.4, 95% CI 0.8 to 2.6, p=0. 3) is not significantly different.8
The aetiology of PTB is variable. Approximately one-third of PTBs are iatrogenic, often a result of adverse events (AEs) such as pre-eclampsia, eclampsia or intrauterine growth restriction.9 Spontaneous PTB accounts for the other two-thirds of PTB; however, in the majority of the cases, the causative reasons remain elusive.10 A number of proxies for spontaneous PTB have been identified, such as genital tract infections, inherited predisposition, uterine ischemia and over distension, decline in progesterone action, disruption of maternal-fetal tolerance, cervical dysfunction/weakness or endocrine disorders, such as gestational diabetes mellitus (GDM), and changes in the placental microbial composition, which can lead to the production of proinflammatory cytokines and subsequent inflammation.11–15 Tropical communicable diseases (eg, malaria and tuberculosis) are more prevalent in LMICs and have been identified as a contributor to the burden of PTB in these settings.16–18
It is generally accepted that PTB results from heterogeneous influences that can lead to preterm labour or premature rupture of fetal membranes with subsequent uterine contractions. The current understanding of preterm labour is that due to a shift in signalling pathways, the chain of events causing the myometrium to switch from a quiescent to a contractile state is triggered too early. Priming leucocytes in peripheral blood and leucocyte accumulation in uterine tissue seem to play a key role in the activation of this pathway.19–21 While in term deliveries this process is activated physiologically, in preterm labour external, environmental and internal influences can lead to alterations in the composition of immunomodulating agents and activate a pathway, which Romero et al 22 referred to as ‘common pathway’, ultimately leading to preterm labour.
In the event of microbial-induced preterm labour, bacteria can gain access to the amniotic cavity either via an ascending route (eg, from the lower genital tract) or a haematogenous route (eg, microbes from the oral cavity); an abnormal vaginal microbiota or vaginal dysbiosis is known to be associated with PTB.13 23 Bacterial vaginosis is a known risk factor for PTB and other adverse pregnancy outcomes.24 25 The vaginal microbial composition changes over the course of pregnancy but whether the vaginal microbiome is altered when comparing microbial composition between pregnant women delivering at term versus preterm is not clear.26–28 Similar to vaginal microbial composition, the composition of the intestinal microbiome undergoes changes during pregnancy.29 30 Whether there are implications of intestinal dysbiosis on pregnancy outcome is not yet known.
A successful pregnancy relies on complex molecular and biochemical processes that are essential for the differentiation, development and preservation of placental tissue as well as fetal development and maturation.31 32 To ensure adequate functions of the placenta, genes are upregulated and downregulated adaptively depending on progress of the pregnancy and the exposure to environmental stressors.32 In analogy to gene regulation, levels of placental DNA methylation and post-translational modification of histones, an epigenetic process that does not involve modification of the DNA sequence, are significantly altered in suboptimal maternal conditions, such as GDM, pre-eclampsia and obesity.33–35 Technological breakthroughs in the field of high throughput molecular profiling technologies enable assessment of these biochemical processes by simultaneous measurement of abundance or activity of all elements constitutive of a given system (genome, transcriptome, proteome, microbiome and so on).
Molecular signatures are sets of genes, proteins, genetic variants or other variables that can be derived from systems-scale profiling and used as markers for a particular phenotype.36 These so-called systems approaches have revolutionised biomedical research by enabling the phenotyping of individuals and their environment at unprecedented depth and play a pivotal role in establishing personalised medicine.37
Approaching the dilemma of PTB with a structured systems approach that builds on monitoring perturbations in blood transcriptome and human microbiome profiles during pregnancy might enable observations of significant changes in molecular signatures peripartum and has the potential to gear researchers and clinicians towards a predictive tool for risk assessment of PTB in the early stages of pregnancy.38 39 To accomplish this, we aim to obtain holistic multiomics data on preterm delivery by integrating transcript abundance (the expressed fraction of genes of a genome) and microbial composition of the gut and vaginal microbiome with multilevel data of placental tissue and cord blood. Notably, the introduction of an additional dimension with high frequency sampling and profiling is meant to enable the identification of molecular changes underlying pathogenic processes prior to onset of symptoms. This so-called molecular interception approach is meant to provide novel avenues for the development of preventive strategies aiming to interrupt such processes and avert the adverse clinical outcomes.
Methods and analysis
Study design
This trial is designed as non-interventional prospective cohort study in pregnant women without severe medical or obstetric complications to assess potential differences in whole blood transcript abundance and changes in gut, vaginal and oral microbial composition from the first trimester until 12 weeks postpartum associated with PTB. In addition, bacterial composition as well as RNA profiles from placenta and cord blood serum will be studied.
Setting
This trial is carried out as cooperation between Shoklo Malaria Research Unit (hereafter referred to as SMRU), Mae Sot, Thailand, and Sidra Medicine (hereafter referred to as Sidra), Doha, Qatar. SMRU is a field station of the Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand, and is part of the Mahidol-Oxford Research Unit. For more than three decades, SMRU has contributed to and combined health services and research in rural and disadvantaged migrant and refugee populations living on the Thai–Myanmar border. SMRU recruits study participants alongside the Thailand–Myanmar border and collects clinical data and study related samples. Sidra is part of Qatar Foundation for Education, Science and Community Development. The Research branch of Sidra serves as a hub for biomedical research in Qatar and aims to understand mechanisms of disease aiming to develop modern tools to diagnose disease and improve health outcomes of women and children. Sidra analyses samples as described in the methods section below.
Participants and recruitment
In total, 400 study participants will be recruited at SMRU antenatal care (ANC) clinics on the Thailand–Myanmar border. All eligible pregnant women will be approached and informed about the background of the study in their preferred language by a trained counsellor. Gestational age will be estimated by early ultrasound scan by locally trained technicians40; subjects with an estimated gestational age from 8+0 to less than 14+0 (weeks+days) weeks will be included. Inclusion and exclusion criteria are summarised below.
Inclusion criteria
Pregnant woman willing and able to give informed consent.
Karen or Burman ethnicity.
Age 18-49 years.
Healthy, with viable singleton first trimester (8+0 to <14+0 weeks) pregnancy.
Planning to deliver at SMRU clinic.
Able (in the Investigators opinion) and willing to comply with study requirements.
Exclusion criteria
Emergency obstetric care required.
Pregnant woman with medical or obstetrics complications that would make it difficult to comply with study requirements (in the investigator’s opinion).
Sample size
Since the number of samples necessary for the identification of a robust signature is variable, sample size requirements will depend on the amplitude of the difference between and the variability within study groups. Best practices in the microarray field dictate utilisation of at least two independent sets of samples for the purpose of validating candidate signatures (or profiles).41 However, in the absence of prior high-resolution temporal profiling data in this setting, a pragmatic approach was chosen to determine the samples size: recruitment of a number of cases and controls from which a candidate biomarker signature with potential clinical relevance can reasonably be expected to be found. The number of cases was set to 30, which means that we consider it unlikely that a signature would be sufficiently robust to be of clinical relevance if it required a higher number of cases and controls to reach significance. This number is selected empirically, but our experience conducting blood transcriptomic studies across a wide range of study settings indicates that it would likely be sufficient to identify candidate signatures and assess phenotypic heterogeneity at the molecular level.42
With a target number of cases being set (in our case to 30), the estimation of the cohort size (n=400) is then simply based on documented PTB rates for the study population, which is approximately 8%. The rate of PTB in this population is on the lower end of the spectrum, and we also considered the risk that, as is the tradition, some women deliver at home whereas the study needs to capture unit births.
Study and sampling procedures
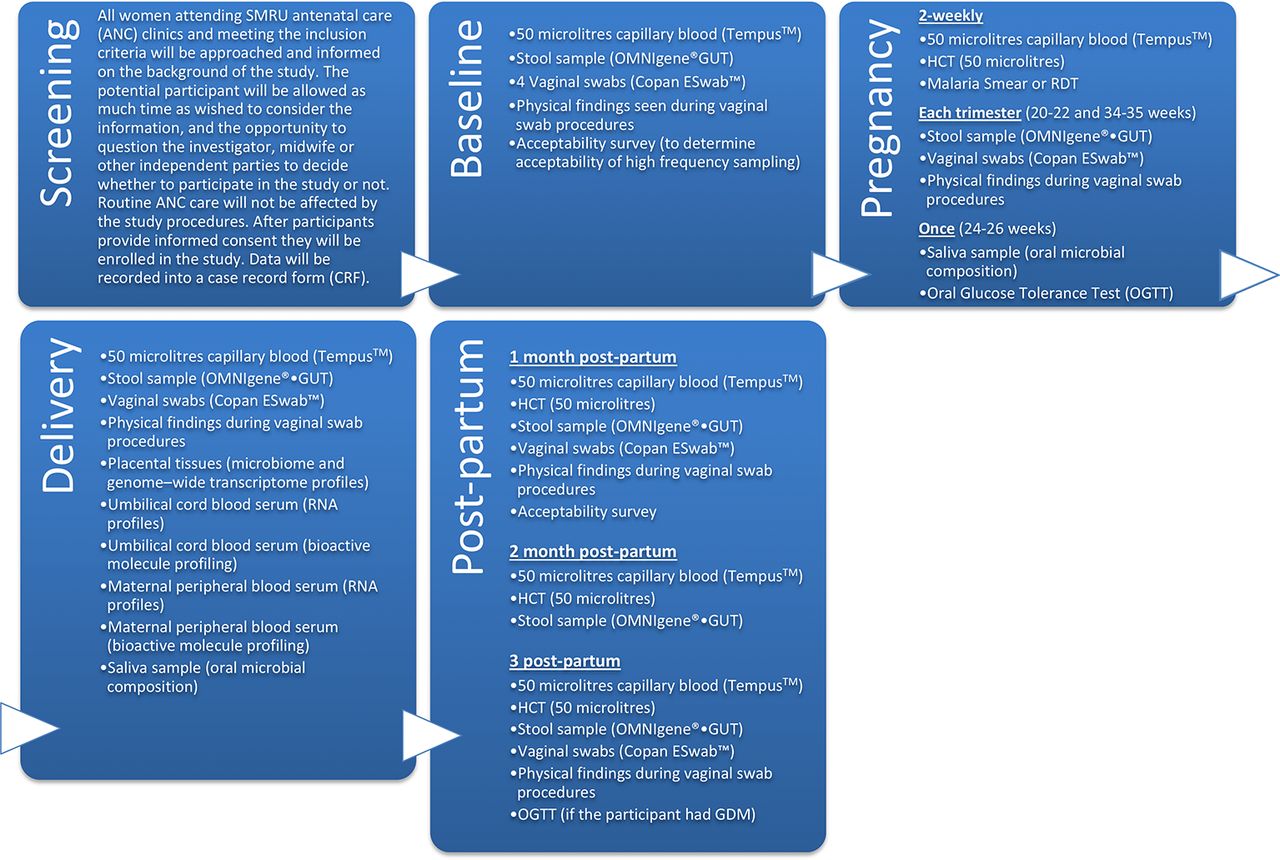
Table 1 as well as figures 1 and 2 provide an overview of the study conduct and timing regarding patient related data, assessment and samples; details on routine ANC procedures are provided in the online supplementary file.
Supplementary file 1
Study timeline and conduct

Timeline of study procedures. OGTT, oral g lucose tolerance test

{kind=link}
{kind=link}
Study-specific procedures. SMRU, Shoklo Malaria Research Unit.
Below is a brief narrative of samples taken to achieve study objectives.
To obtain RNA for gene expression data, small whole blood samples (50 µL) are collected via finger prick sampling into a capillary straw and mixed with a RNA stabilising solution.43 44 Total RNA is extracted, and the transcriptome signatures of mothers with term and preterm infants are compared.45
To assess the vaginal microbiome, vaginal swabs are collected from the posterior fornix. Four vaginal swabs are taken each time: (1) to extract genomic bacterial DNA, (2) to prepare a Gram stain smear to assess the Nugent score,46 (3) for metatranscriptomic analysis and (4) for analysis of vaginal cytokines.
Fresh stool samples are taken and transferred into a DNA stabilising solution for analysis of microbial composition. A 24-hour food recall is asked each time a stool sample is provided to correlate dietary intake with intestinal microbial composition.
To evaluate the microbial composition of the oral cavity, saliva samples are taken twice.
To assess potential microbial colonisation in the fetoplacental compartment, placental tissue as well as cord blood and maternal blood samples are harvested at delivery. RNA profiles (mRNA, long non-coding and short non-coding) will be established from the placenta tissue. Serum from cord blood and maternal blood at delivery will be used to isolate circulating RNA (long non-coding and short non-coding) as well as to determine the concentration of hormones such as adrenocorticotropic hormone (ACTH), cortisol and cytokines.
In the event of a febrile episode, a set of capillary blood, stool and vaginal swab samples is repeated. Moreover, a standard fever screening battery (available in the online supplementary file) is triggered, and clinical and treatment information (eg, antibiotic use) will be captured. Whether high frequency sampling is accepted will be assessed by a survey conducted on enrolment and after delivery.
After the postpartum period, the researchers will follow the mothers and infants for a total period of 2 years to build a mother–infant cohort. Rationale for following the progeny and exact procedures will be shared in a separate publication of the respective research protocol.
Primary outcome measures
The primary outcome is the characterisation of the molecular signature of women with a preterm delivery. In a nested case–control analysis of the study cohort, these cases are then compared with a control group of women with a term delivery and an uneventful pregnancy, matched by parity, maternal age, ethnicity, first trimester body mass index and smoking status.
Secondary outcome measures
Identified biomarker signatures predictive of poor fetal growth.
Describe the variation in molecular signatures during pregnancy and postpartum in:
Individual women (intrapregnancy).
Between women (interpregnancy).
Determine the acceptability of frequent sampling in the study population.
Preliminary data on the potential of a system approach in detecting infectious events before the onset of clinical symptoms or in the absence of clinical symptoms.
Methodologies of sample and data processing
Blood transcriptional profiling
Blood transcript abundance will be measured via RNA sequencing and a high-throughput quantitative PCR platform and assayed through ‘transcriptome fingerprinting’.
RNAseq
This will be carried out by the Sidra genomics facility. Libraries will be constructed from globin-reduced RNA using the Illumina TruSeq RNA Sample Preparation kit according to manufacturer’s instructions. Libraries will then be clustered on a flow-cell using the TruSeq Single Read Cluster Kit followed by paired-end sequencing of 150 nucleotide fragments on a HiSeq2500 sequencer (Illumina) with targeted read depth of 20 M reads/sample. RNAseq alignment and quality control will be carried out by the Sidra bioinformatics core.
Transcriptome fingerprinting assay
A manuscript describing this assay is in preparation. Briefly, a collection of 985 blood transcriptome profiles spanning 16 different diseases (autoimmune, infectious, cancer and genetic) or physiological states (pregnancy) will be used as a basis for the identification of 382 gene sets (modules) capturing a wide repertoire of transcriptional signatures. The gene sets will be further grouped in 38 classes, from which 66 representative modules will be selected. Four representative genes/module will be subsequently selected, resulting in a set of 264 target and 8 housekeeping transcripts used as a basis for targeted assay development. In this study, abundance of these 272 transcripts will be measured on a Fluidigm biomark high throughput PCR instrument. Cycle threshold (Ct) values will be generated and converted to fold changes (FCs) to reference samples. First, delta Ct values will be calculated by subtracting the geometric mean of the housekeeping genes to control for different starting amounts, then delta-delta Ct values will be calculated by subtracting the geometric mean of the reference sample to control for potential plate batch effects, and finally FC values will be calculated by taking 2 to the power of the negative delta-delta Ct.47 48 Module FC values will be calculated as the geometric mean of the FC values of the four genes in the module.
Microbial nucleic acid isolation and 16S rRNA gene sequencing (all samples intended for microbiome analysis)
Microbial genomic DNA will be extracted from the stool samples and vaginal swabs using MoBio Power Fecal isolation Kit and the MoBio Power Soil isolation Kit (MoBio Laboratories, Carlsbad, California, USA) respectively, from saliva samples using automated extraction with QIAsymphony DSP DNA kit on QIAsymphony (Qiagen, Germany) according to the manufacturer’s instruction. Samples will be stored at −20°C until sequencing. The quantity of purified DNA will be measured at 260/280 absorbance using a NANODROP spectrophotometer (Thermo Scientific), and the quality will be evaluated by electrophoretic fractionation in 0.5× TBE gel at 0.5% agarose (w/v), stained with Midori Green and will be recorded using a Bio-Rad Molecular Imager Gel DOC XR+ Imaging System.
The V1–V3 regions of the 16S rRNA will be amplified from microbial genomic DNA using the different forward primer: 27F with 12 base pairs (bp) golay barcodes and Illumina 5′ adapter for each sample and common reverse primer 534 R.49 In brief, PCR will be performed in triplicate in a 50 µL reaction mixture containing 10 ng of template DNA and 2× KAPA HiFi HotStart Ready Mix. The following thermal cycling conditions will be used: 5 min of initial denaturation at 94°C; 30 cycles of denaturation at 94°C for 30 s, annealing at 62°C for 30 s and elongation at 72°C for 30 s; and a last step at 72°C for 10 min. The amplified PCR products of approximately 625 bp in size from each sample will be pooled in equimolar concentration. This pooled PCR product will be purified through E-Gel SizeSelect Agarose Gels Starter Kit, 2% (Thermo Fisher Scientific). High-throughput sequencing will be performed on an Illumina MiSeq 2×300 platform (Illumina, San Diego, California, USA) at Sidra in accordance with manufacturer’s instructions. Image analysis and base calling will be carried out directly on the MiSeq.
Demultiplexed sequencing data will be analysed using QIIME software V.1.9.0 pipeline.50 FASTQ files will be converted into FASTA files, and all demultiplexed files will be concatenated into a single file. Further analysis will be performed as previously reported.51 52 Sequence alignments will be done against the Greengenes core set.53
Alpha diversity will be measured by R software, using the phyloseq package (1,2).54 Beta diversity will be represented using weighted UniFrac distance measure, and contributions to the differences in the beta diversity will be presented as principle coordinate analysis as proposed in QIIME.55
Placenta and blood serum transcriptional profiling
Total RNA and long non-coding RNA will be isolated from 30 mg of frozen placenta tissue using the RNeasy kit (Qiagen) or from 100 µL serum using the RNeasy Serum/Plasma kit (Qiagen) while short non-coding RNA will be isolated from 30 mg of frozen placenta tissue using the miRNeasy kit (Qiagen) or from 100 µL serum using the miRNeasy Serum/Plasma kit (Qiagen). Libraries will be prepared using TruSeq RNA Library Prep kit (Illumina) according to the manufacturer’s instructions and RNAseq using the Illumina HiSeq 4000 platform. Paired-end reads (300 cycles) will be performed. Sequence data quality will be examined using the FastQC tool, followed by the reads mapped to the human reference genome (hg 19).
Detection and quantification of bioactive molecules
Serum (originating from cord blood or maternal blood at birth) levels of ACTH, cortisol and cytokines will be tested using commercially available ELISA kits (R&D) according to the manufacturer’s instructions.
Statistical analysis
Descriptive data
Appropriate statistical tests will be applied to analyse and present descriptive data.
RNASeq data
The data in *.fastq files will be assessed to investigate the read quality and experimental artefacts including sample contamination, PCR and sequencing errors and so on. Sequence alignment will then be performed, reads will be mapped to the human reference genome (hg19) and quality will be reassessed. Postalignment, reads will be deduplicated, quantified to identify expression level of genes/transcripts and normalised according to a suitable method. Genes will be filtered based on their normalised signal intensity values with a specified cut-off.
The statistical environment R will be used to perform statistical analysis along with the PartekR FlowR. Appropriate statistical test will be employed to identify genes showing statistically significant differences with p value cut-off ≤0.05 and multiple testing correction (FWER/FDR) method to control false positives and false negatives. The FC in gene expression will also be measured. The set of differentially expressed genes identified by measuring FC difference and/or using p<0.05 cut-off will be retained and used further for the downstream analysis including Gene Ontology (GO), Gene Enrichment and Pathway Analysis. Hierarchical clustering will be performed to investigate the expression profiles in the datasets. Transcriptional modular analysis will also be performed as described previously.56 For the longitudinal pregnancy data, mixed effects models will be run using the lme4 package in R to compare modular trajectories of uncomplicated pregnancies and preterm.57
Microbiome analysis
Alpha diversity measures such as Observed, Chao1, Shannon and Simpson indices will be analysed using minitab17 (Minitab statistical software). Kruskal-Wallis tests will be used to compare the statistical difference among diversities between the categories. P values lower than 0.05 will be considered statistically significant. A multivariate analysis between the relative abundance of microbiota and clinical metadata will be done using MaAsLin.58
Multiomics analysis
Exploratory and unsupervised data analyses of single omics datasets will be performed along with principal component analysis to capture trends/variations in the datasets. Then, the different omics datasets will be integrated and compared with understand some of the complex steps and processes using correlation analysis, regression analysis, meta-data analysis and pathway analysis. Correlation analysis will be used to measure the strength and directionality of pairwise relationships between different datasets. Metadata analysis will be used to visually depict sample attributes such as phenotype, molecular subtypes, clinical parameters and time points. Venn diagrams will be used to display the overlap between different omics datasets. An integrated pathway analysis will also be performed with statistically significant variables identified from single omics datasets. Based on the significant variables, a prediction model will be built for monitoring of pregnancy complications.
Patient and public involvement
Historical data from monitoring pregnant women in this area show that PTB is problematic due to the difficulty in accessing high-level neonatal care and due to the association with neonatal death. Study participants were not directly involved in the study design, the elaboration of research questions and outcome measures or the recruitment and conduct of the study. However, the protocol was reviewed and approved by the local Tak Province Community Ethics Advisory Board (T-CAB). T-CAB members are representatives of the local community who act as a bridge between researchers and the local population.59 Their role was to advise on ethical and operational aspects of the study (eg, appropriate informed consent procedures, compensation and confidentiality), and they will provide a mechanism to inform the community of the results of this study. Additionally, the researchers will involve study participants to assess whether the study procedures and frequency of sampling is accepted by conducting acceptability surveys at study enrolment and study completion.
Study results that can positively change clinical practice will be disseminated through the local annual health meeting and, if relevant, by health education at the antenatal clinics. Individual data will not be reported back to participants.
Ethical considerations
The population served by SMRU clinics is predominantly migrant workers from Myanmar living alongside the Thailand–Myanmar border. SMRU has been working with the border communities for over three decades, a long experience of working with vulnerable populations. SMRU medics, nurses, midwives and health workers are recruited from the same areas as the patient population and are sensitive to their needs. All communication is conducted in the preferred language of the patient.60 Trained counsellors will provide informed consent once a pregnant woman has a confirmed viable singleton pregnancy and after routine enrolment procedures for ANC are completed. The participant must personally sign and date the latest approved version of the informed consent form (ICF) before any study-specific procedures are performed. The participant information sheet and ICF detail no less than the exact nature of the study and the implications and constraints of the protocol and the known side effects and any risks involved in taking part. It will be clearly stated that the participant is free to withdraw herself from the study at any time for any reason without prejudice to future care and with no obligation to give the reason for withdrawal.
Written informed consent will then be obtained by means of participant dated signature and dated signature of the person who presented and obtained the informed consent (or thumbprint in the case of non-literate participants).
Confidentiality and access to data
Confidentiality and access to data are according to Standard Protocol Items: Recommendations for Interventional Trials.61 All study-related information is stored securely at the study site. Participant information is stored in locked file cabinets in areas with limited access. To maintain participants’ confidentiality, all laboratory specimens, reports, collected data and administrative forms are identified by a coded ID number only. The participants are assigned a unique study ID, led by the study acronym MSP (eg, the first participant will be coded as MSP-001). All records that contain names or other personal identifiers, such as locator forms and ICFs, are stored separately from study records identified by code number. All study data are entered into an online MACRO database for later analysis. The database is secured with password-protected access systems. Forms, lists, logbooks, appointment books and any other listings that link participant ID numbers to other identifying information are stored in a separate, locked file in an area with limited access.
Only SMRU staff with explicit permission has access to the patient-related data. The data will be made available to colleagues and peer reviewers on request. Variables containing sensitive information may be removed before sharing datasets.
Once the study is completed, anonymous data will be kept at SMRU and securely stored for 10 years.
Data monitoring
The principal investigator designated an independent monitor to perform a review of ongoing study progress and safety. A data monitoring committee is not required for an observational study design.
Spontaneously reported AEs are recorded, and severe adverse events are reported to the local ethical committee. No interim analysis will be carried out and no audit is expected during the conduct of the trial.
Dissemination
The results of this study will be published in international peer-reviewed scientific journals. This project is cooperative, in that each participating principal investigator will take the lead in publishing results relating to his or her respective clinical or ‘omics’ component. It is also collaborative in that integrative analyses across these components will be subsequently carried out collectively and published. Hence, the researchers anticipate multiple publications staggered over time.
Data sharing
Due to ethical and security considerations, the data that support the findings in this study will be accessible only through the Data Access Committee at Mahidol Oxford Tropical Medicine Research Unit. The data sharing policy can be found here: http://www.tropmedres.ac/data-sharing. The application form can be found in the online supplementary file. Transcript profiling data will be shared on the NCBI Gene Expression Omnibus (GEO). The 16S rRNA sequencing data generated from the various microbiome analyses will be stored as a bioproject with a unique ID in NCBI for public resource as Sequence Read archives. (https://www.ncbi.nlm.nih.gov/bioproject/).
Discussion
The aim of this study is to identify potential predictive ‘molecular signatures’ associated with PTB as well as maternal and neonatal morbidity and mortality in a resource-limited setting where infectious diseases such as malaria and tuberculosis are more prevalent. Adverse pregnancy outcomes like prematurity are important phenotypes that significantly influence neonatal morbidity and mortality, while also impacting life beyond the neonatal period.4 5 Emergence of PTB is poorly understood, and the causative factors often remain elusive. Among identified risk factors are genital tract infections, inherited predisposition, uterine ischaemia and over distension, decline in progesterone action, disruption of maternal fetal tolerance, cervical disease or endocrine disorders and changes in the placental microbial composition and subsequent inflammation.11 12 14 15 GDM, which is also associated with iatrogenic PTB, is an emerging concern in the study population as well as worldwide and is therefore a considerable factor.62–64
To address these various components, we attempt to characterise the phenotype ‘preterm’ by integrating multilevel omics profiles commencing in early pregnancy through to delivery. High throughput technologies can capture a wide array of genomic and environmental parameters of individuals and play a pivotal role in establishing a personalised medicine approach.37
Rapid technical advances improved the quality and quantity of high-resolution molecular profiling and enable to capture of the diversity of a person across a wide array of parameters and, not least, improving cost-effectiveness of sample analysis.65 Heng et al showed that measurement of whole blood gene expression at two time points during pregnancy has the potential to predict spontaneous PTB when combined with clinical data.39 However, the current study is uniquely geared to investigate potential clinical utility of ‘molecular interception’ approaches due to the increased sampling frequency. Genome-wide transcript signatures at an increased sampling frequency will be employed to evaluate whether changes in blood transcript abundance trajectories precede preterm deliveries and can be reproduced in different populations.
Due to the complexity of PTB, perturbations of gut and vaginal microbiome will be incorporated in the analysis, both of which are influenced by pregnancy. The vaginal microbiome is altered when comparing pregnant and non-pregnant women.66 67 Moreover, dysbiosis in the vaginal microbiome could be associated with adverse pregnancy outcomes and potentially enable the identification of pregnant women at risk for PTB at an early stage in pregnancy.25–27 Analysis of the vaginal microbiome is included in this study to characterise the vaginal bacterial composition prepartum, intrapartum and postpartum in a study population which, to our knowledge, has not been done before and to analyse whether perturbations in this microbial niche are associated with preterm delivery. Corresponding to vaginal microbiome, the intestinal microbial composition also undergoes changes during pregnancy.29 68 While data on the association between PTB and changes in the intestinal microbiome are scant, there is some evidence that the gut microbiome is different in mothers with preterm infants.69 Assessment of gut and vaginal microbiome in the 3-month postpartum period will inform about adaption of bacterial composition following childbirth. Since there is a link between PTB and periodontal disease, association of the oral and placental microbial flora will round up this holistic approach aimed at unravelling the dilemma of PTB.23
We anticipate that this project that will tag onto routine ANC visits, will be feasible and acceptable and can demonstrate the potential for the use of a ‘systems approach’ in the prediction of adverse outcomes and complications in pregnancy and early infancy in rural tropical settings. The ultimate goal is to identify predictors of PTB that can guide the most effective targeted intervention strategies for women at risk for PTB in the future. This project is even more relevant to the resource poor population that SMRU is working with because the burden of mortality and morbidity from PTB is significant when only limited neonatal care can be provided.
Since only a fraction of the pregnant women in this cohort are necessary to achieve the case–control comparison of molecular signatures in term and preterm pregnancies, data will be captured from other phenotypes (eg, birth weight, small for gestational age or subclinical malaria infection). Depending on the quantity and quality of the captured data, valid results from any subgroup analysis will be made available for the scientific community and published in peer-reviewed journals.
The following attributes reflect the strength of this protocol:
The prospective study design by building a pregnancy–delivery cohort.
The nature of the proposed systems approach, a holistic approach in which investigators do not need to choose the parameters that will be measured as it will by default simply be ‘everything’, and it is in that sense that these approaches are inherently unbiased.65
High-frequency sampling throughout the entire pregnancy, facilitated by enrolment in the first trimester, which is ensured by precise estimation of gestational age via early ultrasound scan for all participants, enables to evaluate transcript abundance in the peripheral whole blood that contains granulocytes and mononuclear cells in a genome-wide scale.65
Concerted assessment of the interdependence between the elements of blood transcriptome and the microbiome component will enable drawing inferences on how they mutually influence each other and what the consequences for pregnancy outcomes are.
Expected favourable acceptability of high-frequency small volume finger prick capillary blood sampling due to decades of regular malaria (1 to 2 weekly) screening at ANC clinics at SMRU as the only means of effective control in pregnancy.70 Whether high-frequency sampling with the limitation of lack of personal benefit is also accepted will be assessed by acceptability surveys at study enrolment and study completion.
Potential recommendations on sampling time points and frequency for further studies depending on output of this protocol.
Potential limitations of the study protocol:
Selection and exclusion bias: women with presumably complicated pregnancies or women who will potentially require a caesarean section (eg, history of caesarean section) are excluded from the study since SMRU cannot provide advanced medical services due to the rural setting and access to clinic-based but only limited access to hospital-based facilities. Dependent on the medical and obstetric history, case-by-case decision by the attending obstetric doctor and/or research physician whether a potential participant can be enrolled.
Protocol deviations due to the high-frequency sampling: it is expected that some women will not be able to provide all per-protocol samples.
Vaginal swabs at delivery will be dependent on the timing when the pregnant woman arrives at the clinic. Late presentations and/or precipitate deliveries might lead to loss of delivery swab samples.
Loss of participants: despite the long history of work with the local population of more than 30 years and since the majority of SMRU staff originate from the local population, both of which are likely to enhance the operational feasibility of this ambitious project, loss of participants is also expected since the study is conducted in a mobile migrant population. For this reason, women who are lost before birth outcome will be replaced by recruitment of a new participant in line with the sample size calculation.
Epidemiological and technical biases that are associated with gene expression profiling (eg, gene panel selection bias or technical bias such as sequencing bias in library construction or amplification bias).
In conclusion, should this study deliver significant results that are reproducible in other study populations, molecular signatures based on a cross-omics approach could be a potential tool to predict preterm deliveries in the future and allow early/programmed interventions to alleviate morbidity and mortality from these deliveries. Moreover, we might gain more insight in the origin of preterm deliveries that cannot be diagnosed at this time point.
Acknowledgments
The Shoklo Malaria Research Unit is part of the Wellcome Trust Mahidol University Oxford Tropical Medicine Research Programme supported by the Wellcome Trust of Great Britain (Major Overseas Programme – Thailand Unit Core Grant: WT-106698). Sidra Medicine is a member of the Qatar Foundation for Education, Science and Community Development.
References
Footnotes
Contributors DC, RM and FN conceived the overall idea of the project with contributions on the details from SAK, AT, AKM and BSAK. TB, RM, DC, AT, SAK, TK, MT and DHP wrote the manuscript. TB and SS will collect the data. TB, DC, BSAK, SAK, AT and AKM will analyse the samples. TB, DC, BSAK, MT, SAK, AT, AKM, AA, JCK, SL, SP, RM, JU, DHP and FN will interpret the data, write the manuscripts and disseminate results. All authors read and agreed to the final version of the protocol and the publication of the protocol.
Funding Sidra Medicine is the funder of this trial.
Competing interests None declared.
Patient consent Not required.
Ethics approval This research project was approved by the ethics committee of the Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand (Ethics Reference: TMEC 15–062), the University of Oxford Central University Research Ethics Committee (Ethics Reference: OxTREC: 33–15) and the local Tak Province Community Ethics Advisory Board.
Provenance and peer review Not commissioned; externally peer reviewed.