Article Text

Abstract
Introduction Therapeutic schedules for treating neonatal seizures remain elusive. First-line treatment with phenobarbital is widely supported but without strong scientific evidence. Levetiracetam (LEV) is an emerging and promising antiepileptic drug (AED). The aim of this phase II trial is to determine the benefits of LEV by applying a strict methodology and to estimate the optimal dose of LEV as a first-line AED to treat seizures in newborns suffering from hypoxic-ischaemic encephalopathy.
Methods and analysis LEVNEONAT-1 is an open and sequential LEV dose-finding study. The optimal dose is that which is estimated to be associated with a toxicity not exceeding 10% and an efficacy higher than 60%. Efficacy is defined by a seizure burden reduction of 80% after the loading dose. Four increasing dose regimens will be assessed including one loading dose of 30, 40, 50 or 60 mg/kg followed by eight maintenance doses (ie, a quarter of the loading dose) injected every 8 hours. A two-patient cohort will be necessary at each dose level to consider an upper dose level assignment. The maximal sample size expected is 50 participants with a minimum of 24 patients or fewer in the case of a high rate of toxicity. Patients will be recruited in five neonatal intensive care units beginning in October 2017 and continuing for 2 years. In parallel, the LEV pharmacokinetics will be measured five times (ie, 30 min; 4 and 7 hours after the loading dose; 1–3 hours and 12–18 hours after the last maintenance dose).
Ethics and dissemination Ethics approval has been obtained from the regional ethical committee (2016-R25) and the French Drug Safety Agency (160652A-31). The results will be published in a peer-reviewed journal. The results will also be presented at medical meetings.
Trial registration number NCT02229123; Pre-results.
- levetiracetam
- newborn
- seizure
- phase li trial
- hypoxic-ischaemic encephalopathy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
For the first time, levetiracetam (LEV) will be used as first-line treatment for neonatal seizures rather than as an add-on therapy.
The targeted population (ie, newborns less than 3 days old) is particularly vulnerable, and the ethical authority requires the written consent of both parents before LEV administration.
The restricted availability of electroencephalogram (EEG) devices in each investigating centre led after the cessation of seizure burden to a minimal requirement of a 1 hour EEG recording on days 1, 2, 3 and 6.
The statistical model is designed for a rare clinical situation with a sequential adaptive method, which updates in real time the dose allocation for the next patient based on all available data from previous participants.
On average, the proposed design prompts recommendations of the correct dose at approximately 60% of efficacy for a sample size of 30, increasing to over 80% in many scenarios for a sample size of 50.
Introduction
Neonatal seizures occur in approximately 1 to 5 per 1000 live births.1 Hypoxic-ischaemic encephalopathy (HIE) represents the first aetiology of neonatal seizures.1 2 In 80% of cases, seizures associated with HIE occur in the first 3 days of life.2 Seizure treatment during the neonatal period creates issues concerning the developing brain facing deleterious effects of seizure burden (SB) to the potential toxicity of conventional antiepileptic drugs (AED). Van Rooij et al demonstrated that the seizure duration was correlated with brain lesions through MRI.3 Therefore, the World Health Organisation recommended in 2011 that anticonvulsant treatment should be initiated immediately when clinically apparent seizures have lasted for longer than 3 min or in the case of brief repeated seizures. This recommendation was supported strongly but no gradation was attributable due to the lack of scientific evidence.4 Furthermore, conventional AEDs such as phenobarbital (PHB) are employed in an off-label manner in neonates. Epidemiological studies have illustrated that PHB is widely used as the first-line treatment across the world with a lack of consensus concerning subsequent add-on lines of treatment, including phenytoin (PHT), lidocaine, midazolam and other benzodiazepines.5 A meta-analysis from the Cochrane Database concluded in 2004 that ‘there is little evidence from randomised controlled trials to support the use of any of the anticonvulsants currently used in the neonatal period’. This statement was recently reinforced by a systematic review published in 2015, which presumed that ‘there is an urgent need for more evidence-based studies to guide neonatal seizure management’.5 6 This vague position resulted from the lack of well-conducted trials and from the limited available data regarding the efficacy and safety of AED use during the neonatal period. Only one randomised controlled trial was performed focusing on AED efficacy in neonates. In this trial, PHB as PHT precipitated seizure cessation in only 44% of cases in monotherapy. The SB intensity appeared to be inversely related to therapeutic success.7 This restricted efficacy might be explained by the signalling pathway of PHB on GABA receptors, which are paradoxically excitatory in the immature brain before transitioning to their inhibitory function.8 9 Furthermore, recurrent seizures induced an intracellular chloride accumulation, reinforcing the excitatory function of GABA receptors and then creating PHB inefficiency in treating intense SB during the neonatal period.10 Moreover, some concerns about the safety of PHB exist. Experimental data have demonstrated that PHB increased in a dose-dependent manner neuronal apoptosis in the immature brain.11 Therefore, a necessity for new efficient and safe AEDs for newborns has emerged. Levetiracetam (LEV) might be the appropriate candidate to fulfil these criteria. First, LEV exhibits an original means of action by reducing, through the SV2a protein, glutamate release by presynaptic neurons and then regulating the intracellular calcium of postsynaptic neurons through NMDA and AMPA receptors.12 Second, LEV appears to be free of toxicity in relation to the neonatal brain. Experimental data have demonstrated that LEV did not induce neuronal apoptosis in the neonatal brain.13 14 Furthermore, an observational study illustrated that LEV cumulative doses received during the neonatal period were not associated with the probability of subsequently developing a cerebral palsy.15 Third, an intravenous galenic form of LEV is available, allowing for the treatment of non-fed newborns. Fourth, an off-label use of LEV as second-line treatment after PHB has now been widely observed.16 17 Retrospective studies reported LEV use in neonates with various dose regimens and administration schedules such as increasing doses until seizure cessation, similar doses twice a day, or a loading dose with subsequent maintenance doses. Loading doses infused to neonates varied from 10 mg/kg to 60 mg/kg.17–25 The maximal amount of LEV infused in a newborn was 150 mg/kg within a 24 hours period.17 Fifth, to date, the treatment of approximately 445 newborns through LEV has been reported with limited side effects including one case of anaphylactic shock and a rare sleepy state fostered by a simultaneous PHB treatment.19 22 23 26 27 Similarly, LEV efficacy for significantly reducing or eliminating neonatal seizures has been recently estimated at 77% of cases in first-line treatment and 66% of cases in add-on therapy in a cohort of 102 patients from five retrospective studies.28 In this context, it is highly important to determine the benefits of LEV for treating neonatal seizures and to determine the most effective and safest dose of LEV in neonates following a rigorous and prospective methodology. In this study, a phase II trial has been designed to identify the ideal LEV loading and maintenance doses in newborns suffering from HIE. An original approach has been adopted by using LEV as the first-line treatment.
Methods and analysis
Study settings
Patient recruitment will be performed in five French Neonatal Intensive Care Units (NICU) (Angers, Lille, Rouen, Rennes and Tours) beginning in October 2017 and continuing for 2 years. These centres have been selected for their expertise in managing neonatal seizures. The coordinating site for this study is the University Hospital Center of Tours (France). The protocol has been written according to the SPIRIT reporting guidelines.29
Participants
Eligible patients are term newborns with hypoxic ischaemic encephalopathy (HIE) who are less than 72 hours old (figure 1). Briefly, three inclusion criteria have been selected: (i) birth term above 36 gestational weeks and birth weight above 1800 g; (ii) perinatal asphyxia, defined as Apgar score equal to or below five at 5 min, resuscitation required at birth or metabolic acidosis on umbilical arterial blood gas or until 1 hour after birth (pH <7.1, Excess Base ≥16 mmol/L or lactate ≥11) and (iii) neurological impairment in the first 6 hours of life, including consciousness, tone, sucking, archaic reflexes or pupillary alterations.
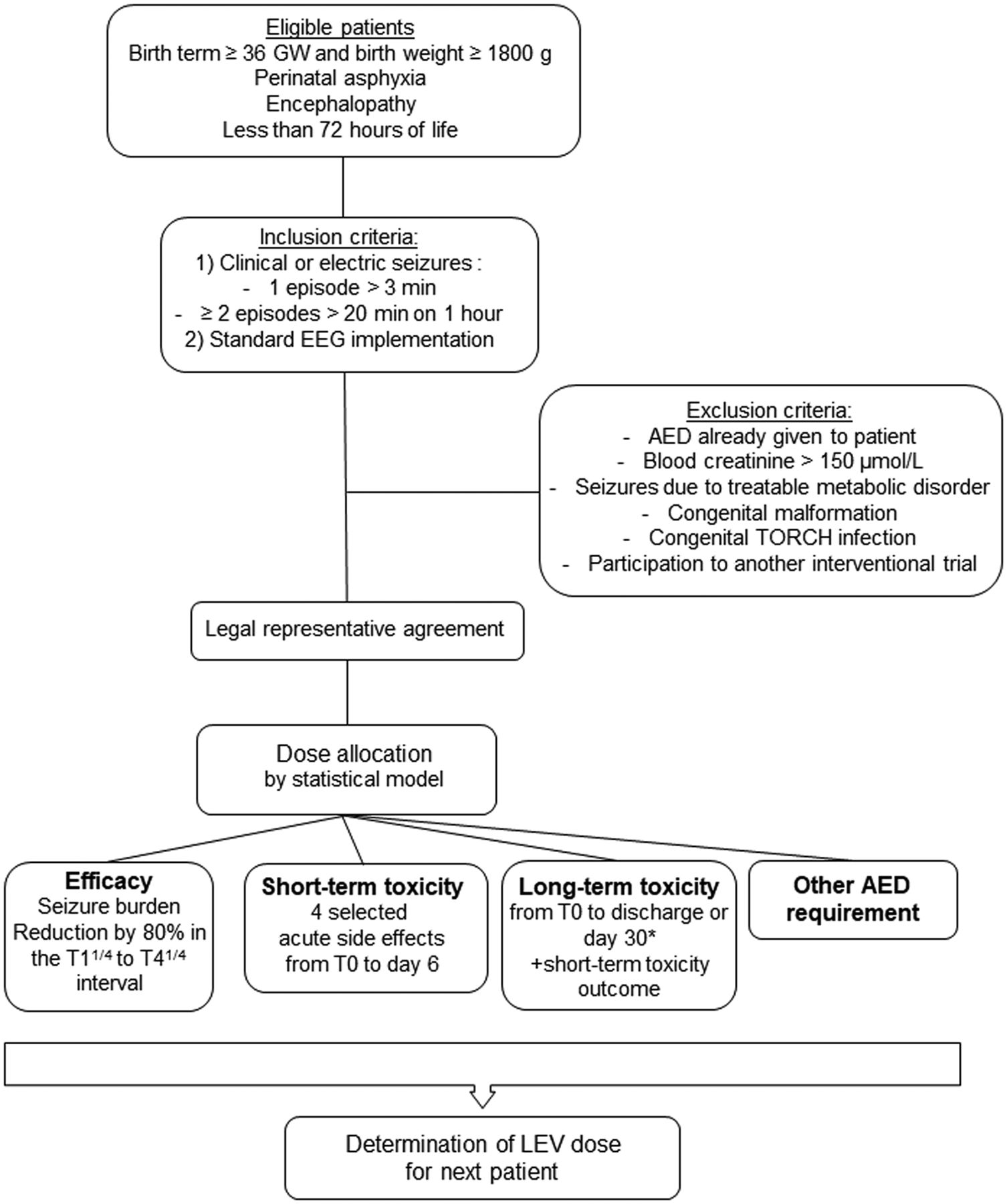
LEVNEONAT-1 study flowchart. AED, antiepileptic drug; EEG, electroencephalogram; GW, gestational weeks; LEV, levetiracetam; T0, levetiracetam loading dose infusion start; T11/4, 1 hour and 15 min after the levetiracetam loading dose infusion start; T41/4, 4 hours and 15 min after the levetiracetam loading dose infusion start.
Brain cooling implementation is delegated entirely to the discretion of investigators according to the French guidelines.30 Therapeutic hypothermia implementation and the rewarming time will be reported in the e-CRF form. Since seizures generally occur during moderate and severe HIE requiring therapeutic hypothermia, it is expected that most participants are on brain cooling. Therapeutic hypothermia is regarded as a variable in the pharmacokinetic analysis.
Inclusion should be considered when clinical signs or an electroencephalogram (EEG) pattern compatible with seizures are recognised by the investigator and when monitoring with a continuous 8-electrode EEG recording is possible. A seizure lasting more than 3 min or more than two seizures lasting more than 20 s within a 1-hour period on a standard EEG recording fulfils the inclusion criteria. Critical-activity recognition on EEG recording is based on the investigator’s experience. A specific training for recognising critical activity on standard EEG has been provided to the investigators in each centre. An inclusion could be diagnosed incorrectly by a retrospective interpretation of an EEG recording by a local electrophysiologist planned as soon as possible (cf. Efficacy Criteria). A second interpretation by an independent and blind reader will be organised in a brief delay to assess the lack of critical activity on the EEG recording. If the lack of critical activity on the EEG is confirmed, the patient will not be included in the statistical model and the experimental treatment will be immediately disrupted, but pharmacokinetic and safety analysis will be performed. Finally, written consent of both parents or authorised guardians and a subscription to social security health insurance are required to complete inclusion.
Exclusion criteria concern patients already treated with an AED aside from a midazolam bolus required for intubation, patients suffering from seizures due to a treatable metabolic aetiology such as hypoglycaemia and hypocalcaemia, patients with severe renal failure associated with serum creatinine above 150 µmol/L, patients with evident signs of genetic or congenital malformations or infectious embryofoetopathy or patients who have already been recruited in another interventional research trial.
Intervention
A therapeutic schedule consists of a loading dose (T0) followed by eight maintenance doses every 8 hours resulting in a 3-day treatment period (figure 2). Eight-hour intervals between doses were implemented based on the LEV pharmacokinetics (PK) obtained from 18 newborns, demonstrating a shorter half-life of 8.9 hours relative to older patients.26 Four increasing loading doses were selected as follows: (i) 30 mg/kg; (ii) 40 mg/kg; (iii) 50 mg/kg and (iv) 60 mg/kg. Each maintenance dose corresponds with a quarter of the loading dose (7.5, 10, 12.5 and 15 mg/kg, respectively). Levetiracetam (levetiracetam Mylan, 100 mg/mL) will be diluted in a 5% glucose solution to a final concentration of 5 mg/mL. LEV will be administered intravenously over a fixed time of 15 min through a central or peripheral line.

{kind=link}
{kind=link}
LEVNEONAT-1 experimental schedule and time-line. AED, antiepileptic drug; AEP, auditory evoked potentials; EEG, electroencephalogram; EFF, efficacy; LEV, levetiracetam; PK, pharmacokinetic; TOX, toxicities.
Principal aim
The principal aim is to determine the most efficient dose regimen under toxicity restrictions of LEV for neonates while accounting for efficacy, toxicity and PK.
Efficacy criteria
Efficacy has been defined as an 80% reduction of SB in an EEG recording between the period immediately before the LEV loading dose (from 20 min to 3 hours) and the 3 hour time interval from 1 hour and 15 min (T11/4) to 4 hours and 15 min (T41/4) after the beginning of loading dose infusion (T0) (figure 2). SB corresponds with the cumulative time of ictal electric activity on the EEG- analysed time lap. A first analysis of the EEG recording will be performed locally at each investigator centre and will be reported in e-CRF format on the sixth day following T0. A second blinded and centralised analysis is scheduled to occur subsequently, every 6 months. If there is more than a 10% difference between EEG interpreters or an opposite conclusion, a third EEG analysis will be performed. A subsequent correction of efficacy criteria in the statistical model can be performed (whenever it is identified). Efficacy criteria will not be accounted for in the dose allocation process in case of a second AED requirement before T41/4 or an unexpected event in LEV preparation or infusion precipitating an unknown dose injection.
Toxicity criteria and safety monitoring
Toxicity will be assessed according to both of the following modalities: (i) short-term toxicity and (ii) long-term toxicity (figure 2). Short-term toxicity has been designed to rapidly trigger a decreasing dose allocation for the next potential participant through an e-CRF alert. Short-term toxicity focuses on four adverse events potentially attributable to LEV: (i) severe apnoea that leads to mechanical ventilation during the 4 hour period following the LEV infusion;19 22 23 26 (ii) anaphylactic shock occurring during the 30 min following the LEV infusion;27 (iii) toxic epidermic necrosis and (iv) Stevens-Johnson syndrome. Investigators must declare the occurrence of one of these adverse events immediately to the pharmacovigilance unit and in the e-CRF. On day 6, if none of these adverse events have been observed, the investigator ticks the ‘no’ box that corresponds to each effect in the e-CRF and short-term toxicity will therefore be regarded as negative. Long-term toxicity encompasses all the adverse events observed and declared to the pharmacovigilance unit up to hospital discharge or the 30th day of life at the latest. A short-term toxicity alert or any serious unexpected suspected adverse reaction (SUSAR) will immediately trigger a meeting of the scientific committee, which consists of a pharmacist (CM) and a neonatologist (GF) and the LEV treatment will be discontinued. If no severe or unexpected adverse reactions are declared, a systematic meeting of the scientific committee will be planned during the 10 days following the participant discharge or the participant’s 30th day of life at the latest. The scientific committee will then determine the imputability (unrelated/possible/probable) and acceptability of each declared adverse event based on the severity at the acute phase, the quality of recovery (partial or complete) with potential subsequent disability and the frequency of occurrence. Ultimately, a single adverse event regarded as imputable to LEV and inacceptable precipitates a declaration of toxicity as positive in the statistical model. The requirement of another AED will also be included in the statistical model as well as the delay between T0 and treatment beginning to the extent that it could alter proper LEV efficacy and toxicity.
Secondary objectives
Pharmacokinetics of levetiracetam: blood samples
LEV PK in participant’s blood will be measured five times at 30 min, 4 hours and 7 hours after the end of LEV loading dose infusion and at 1–3 hours and 12–18 hours after the last LEV maintenance dose (figure 2). Each PK sample requires 500 µL of total blood (ie, 2.5 mL in total). After centrifugation, the plasma will be harvested, and samples will be frozen at −20° or −80° before sending for measurement (VJ). The PK of LEV in the population of the study will be investigated through a population approach.31 The mean values of the PK parameters (elimination clearance, central and peripheral distribution volumes, distribution clearance) and their respective interindividual variability will be estimated. Possible relationships between the covariates (birth bodyweight, gestational age, therapeutic hypothermia) and the interindividual variability of the PK parameters will be investigated. Individual PK parameters will be estimated and used to calculated the maximum concentration and the area under curve (AUC) corresponding with the loading dose, after the first maintenance dose and the cumulative AUC of the entire treatment. Potential relationships between these PK parameters and the efficacy and safety criteria will be investigated, and these pharmacokinetic/pharmacodynamic relationships will be used to determine the optimal dosing regimen.
Seizure recurrence from T41/4 to day 6
Clinical or electric seizures occurrence and frequency during LEV treatment (ie, from T41/4 to T72) and until complete LEV elimination (ie, day 6) will be reported in the e-CRF in addition to concomitant AED treatment. The complete and definitive cessation of seizures will be recorded in the e-CRF. Continuous EEG monitoring is scheduled during the 3-day treatment by LEV as well as a 1 hour recording on day 6. Unfortunately, the restricted availability of EEG devices in each investigating centre after the cessation of SB prompted a minimal requirement of a 1 hour EEG recording on days 1, 2, 3 and 6 after the LEV treatment initiation. Then, the detection of seizure recurrence and the duration of EEG monitoring are then under the responsibility of each investigator.
Pretreatment seizure burden and LEV efficacy
PB and PHT efficacy in relation to complete seizure control have been directly associated with the pretreatment burden seizure intensity.7 Therefore, to explore this association with LEV, a new analysis will be performed retrospectively by adjusting the efficacy criteria to the SB on the pretreatment EEG. Two subgroups will be considered based on the SB intensity on the pretreatment EEG equal to or above 50% of the EEG recording duration (high SB group) and strictly under 50% of the duration (low SB group). LEV efficacy will be deemed positive when a SB reduction of 50% is observed on the post-treatment EEG recording in the high SB group, whereas a reduction of 80% will still be valid for the low SB group.
Patient follow-up
The participant follow-up will continue until hospital discharge or until the 30th day of life. An assessment has been planned that consists of repeated clinical examinations, haemodynamic monitoring, brain-imaging and auditory and electroencephalographic recordings (figure 2). Clinical examinations will be performed at days 1, 2, 3 and 6 through the Thompson score,32 which measures the neurological distress depth. In addition, Amiel-Tison scoring,33 focusing on neurological status of the newborn, is planned to occur on hospital discharge or on the 30th day of life. Arterial pressure and heart rate will be measured immediately before each LEV injection and every 5 min for 15 min, and then every 15 min for 45 min after the LEV injection. Apnoea, bradycardia under 80 beats per minute, and oxygen saturation drops below 85% will be reported. Brain MRI will be performed between the 4th and 8th day of life. An auditory evoked potential measurement will also be required before hospital discharge.
Other AED requirements
If the persistence or recurrence of seizures is observed after the LEV loading dose, the investigators are completely free to initiate another antiepileptic treatment. The drug name, administered dose, therapeutic schedule and treatment duration will be reported in the e-CRF. If another AED is required during the 4 hours following the LEV loading dose ends, the efficacy data will not be included in the statistical model.
LEV treatment cessation rules
LEV treatment will have to be discontinued in any of the following cases: (i) a short-term toxicity or a SUSAR occurs; (ii) serum creatinine raises above 150 µmol/L in the 7–36 hours interval following the LEV loading dose; (iii) a complete unknown LEV loading dose was infused due to a hazardous event; (iv) a mistaken maintenance dose above 60 mg/kg was infused; (v) a limitation of intensive cares begins before the third day of LEV treatment or (vi) at least one of the two parents or authorised guardians withdraws his consent.
Statistical model and dose allocation
LEVNEONAT-1 is an open-label, single group, sequential dose-finding study with four increasing dosage levels. The short-term toxicity, long-term toxicity and the efficacy endpoints were modelled under Bayesian inference. The optimal dose of LEV was defined as the highest efficient dose under the toxicity restrictions. Before the beginning of the trial, efficacy and toxicity thresholds associated with the desirable optimal dose have been selected. The optimal dose should not be associated with less than 60% of efficacy probability and no more than 10% of short-term and long-term toxicity probabilities. After the inclusion of successive cohorts of two patients, the endpoint observations are binarised as follows: efficacy (yes or no), short-term toxicity (yes or no and, if yes, when), long-term toxicity (yes or no), other AED use (yes or no and if yes, when) and the number of infused maintenance doses with timing (figure 1). A statistical model was designed specifically for this trial, because no other dose-allocation method was available for this indication. The model is a sequential adaptive method since it incorporates all the available information before the trial onset and all the data from the trial that have been accumulated for each new cohort inclusion. Based on updated data, probabilities of efficacy, short-term and long-term toxicities are re-estimated after each cohort. The dose allocated to each further cohort is the estimated as the optimal dose known thus far. The first cohort of patients will receive the lowest dose level, and doses will be increased one-by-one based on the model estimates (no dose-skipping will be allowed if the dose was not yet evaluated). Moreover, since long-term toxicity will take an extended time to be observed, a time to event approach will be considered to avoid ceasing inclusions between two successive cohorts.
When a short-term toxicity alert occurs, a reduction of the current loading dose allocated to the lower level is planned until the scientific committee’s conclusion concerning LEV imputability or lack thereof. The maximal sample size is expected to be 50 participants with a minimum of 24 patients. However, the trial will be terminated prematurely if all doses do not reach the efficacy threshold or the lowest dose exceeds the toxicity threshold. When a patient is eligible, the current dose regimen is available on the trial sponsor’s web site. This dose will be renewed in real time according to the previous participants’ data.
Independent data-monitoring committee
An independent data-monitoring committee (DMC) has been established, which includes a neonatologist, a neuropaediatrician and a pharmacologist. A DMC opinion concerning the trial continuation will be solicited every six patients or in the case of an emergency on the request of the scientific committee.
Trial interruption criteria
Three criteria have been identified for trial interruption: (i) a high probability of incorrect dose range (either for efficacy or for toxicity) will cause a temporary interruption of the trial. After the IMC consultation, a new range of doses could be proposed; (ii) new valid information is published during LEVNEONAT-1, which addresses the principal aim and render this trial outdated or (iii) the scientific committee can decide to terminate the trial at any time if an unacceptable toxicity is assigned to LEV.
Patient and public involvement
Patients and the public were not involved in the design of the study.
Ethics
LEV cannot be infused prior to obtaining the written parental or authorised guardian’s consent. One of two parents or authorised guardians can withdraw their consent at any time, prompting the interruption of the newborn participation in LEVNEONAT-1. Safety monitoring would still be performed to ensure adequate treatment of potential LEV side effects but it will not be recorded in the database. An authorisation from parents or authorised guardians will be necessary to use the data obtained before the agreement withdrawal.
Ethical approval for this study (version 4, 06-06-2017) has been obtained from the regional ethical committee (CPP Ouest 1) under the reference 2016-R25 on the 9 November 2016. The French drug safety agency (Agence Nationale de la Sécurité du Médicament) approved LEVNEONAT-1 (version 4, 06-06-2017) under the reference 160652A-31 on the 5 October 2016.
This trial has been registered on EudraCT (20 February 2014) and on Clinical Trial.gov (1 September 2014). The Eudra CT reference is 2014-000791-26 and the ClinicalTrials.gov reference is NCT02229123. Trial registration data are reported in table 1.
LEVNEONAT-1 trial registration data
Data quality
Standardised LEV prescriptions have been designed in an Excel format. These documents have been joined to the allocation-dose web site. Therefore, the investigator will only fill in the allocated dose and the participant’s birth weight to obtain personalised LEV prescriptions (dilution, quantity, infusion speed, monitoring). For each included participant, all data will be anonymised under a specific code (centre city and number of inclusion), and a personalised file (e-CRF) will be created on the trial sponsor’s web site (https://chu-tours.hugo-online.fr/CSOnline/). Data will be reported in the participant’s e-CRF accessible with a personal code (lead investigator). Lead investigators will only have access to the e-CRFs of their own centre. Only the coordinating investigator (GF), scientific committee members (GF, CM) and data manager (EB) have access to all participants’ data. Data will be checked by faxing original paper documents (drug prescriptions, vital-sign monitorings, biological measurements, EEG interpretations) to the trial sponsor. An agent (EB) will be assigned by the sponsor (ie, Universitary Hospital Center of Tours) for meeting the lead investigators and local research teams regularly, based on the inclusion dynamic. These on-site visits aim to monitor the regular filing of consent forms, the compliance with the protocol and the accuracy of the recorded data from source documents. An audit trigger by the French Drug Safety Agency could be possible at any time during the trial course. Data management have been validated through the MR-001 reference methodology.
Dissemination
Any modifications to the protocol that might impact the conduct of the study or affect patient safety will precipitate a substantive protocol amendment and will be reviewed by the regional Ethics Committee and the French Drug Safety Agency. These substantive changes will be communicated to relevant stakeholders (trial registries, regulatory agencies, investigators). The results of the LEVNEONAT-1 study will be published in a peer-reviewed journal following the Uniform Requirements for Manuscripts Submitted to Bio-medical Journals (http://www.icmje.org/). Publications will be distributed to investigational centres and to all relevant persons or organisations. The LEVNEONAT-1 study will also be presented at relevant national and international medical and scientific meetings related to both of the following elements: (i) methodology and biostatistics and (ii) brain development and seizure treatment during the neonatal period. At the end of the study, a summary of the results will be produced for the non-medical public and will be provided to the participants’ parents on demand.
Time-line
Investigational centres were open from the 21 September 2017 to the 20 October 2017. Patient recruitment was effective from the 20 October 2017 for 2 years. The first enrolment occurred in February 2018.
Discussion
Seizure management during the neonatal period remains elusive, and PHB is not completely efficient in terminating critical activity and is not truly safe for the immature brain. LEV might be promising and more suitable in this condition. However, although LEV is widely used in neonatal care units worldwide, no dose regimen has been clearly established.
LEVNEONAT-1 is a particularly original study using LEV as the first-line treatment and not as add-on treatment after PHB, resulting in purer efficacy and safety data and allowing the possibility of a new therapeutic schedule in neonates.
The other original characteristic of LEVNEONAT-1 is the design of a statistical model allowing for a restricted sample size to determine the optimal LEV dose in neonates by integrating data in real time of each participant. The design performances were assessed through extensive simulation studies. On average, the proposed design prompts recommendations of the correct dose at approximately 60% of efficacy for a sample size of 30, increasing to over 80% in many scenarios for a sample size of 50. Moreover, this method maintains an acceptable number of neonates with toxicities.
The first LEVNEONAT-1 weakness is the targeted population, which consists of newborns with less than 3 days of life who suffer from HIE complicated with seizures. The seizure incidence in the HIE context varies according to studies from 29% to 65% of cases.34–36 This discrepancy might be partially due to the caregiver’s ability to recognise clinical signs of seizure and ictal activity on an EEG recording.37–39 In parallel, the relative urgency to obtain parental consent in this stressful context remains a sensitive issue for investigators. This fact could be a critical point for inclusion because seizures can occur early after birth and therefore require AED treatment in emergency. Both parents should then be informed soon after birth even prior to seizure occurrence, and the reflection period of participants’ guardians could be brief. However, the median time of the first seizures reported in the literature was around 9 to 13 hours of life,34 40 allowing for time for reflection to the parents. The second critical point is the opportunity to monitor the newborns through a standard EEG as soon as seizures are identified with various logistical problems according to each investigational centre, including a variable delay or the inability to implement standard EEG monitoring outside of working hours. Further, continuous EEG monitoring is scheduled during the 3-day treatment with LEV and a 1 hour recording on day 6. The restricted availability of EEG devices in each investigating centre led after the cessation of SB to a minimal requirement of a 1 hour EEG recording on days 1, 2, 3 and 6.
In the case of promising efficacy results, a randomised study should be performed further to confirm the findings.
Protocol version
Issue date: 6 June 2017
Protocol Amendment number: 04
Authors: GF, ES, SZ, CM and EB
Revision chronology
2016-Jul-1 Original
2016-Nov-3 Amendment N°1
At the request of French Drug Safety Agency, details about the procedure triggered by the occurrence of a side effect and the allocation-dose process have been added to the protocol.
2017-May-26 Amendment N°2
At the request of French Drug Safety Agency, the upper dose regimen including a loading dose of 60 mg/kg was withdrawn considering the lack of significant data justifying this dosage in newborns.
2017-Jun-6 Amendment N°3
The upper dose level including a loading dose of 60 mg/kg was validated by the French Drug Safety Agency in light of Venkatesan et al.17
References
Footnotes
Patient consent for publication Obtained.
Contributors GF and ES were responsible for the study design including aims, assessment criteria and intervention schedule. SZ and MU provided statistical expertise in trial design, developed the dose-finding model and conducted the statistical analysis. CM and GF assumed the pharmacovigilance procedure. EB handled the legal aspects and authorisations and helped with implementation. VJ designed the pharmacokinetic analysis and performed the LEV pharmacokinetic measurements. All authors contributed to the refinement of the study protocol and approved the final manuscript.
Funding This work was supported by a grant from the French Ministry of Health (LEVNEONAT-1, 2013, PHRCI 2013 API13/T/065). No financial support from industry took part in the LEVNEONAT-1 funding. Levetiracetam will not be provided by industry. SZ and MU were supported by the InSPiRe (innovative methodology for small populations research) project, which was funded by the European Union’s Seventh Framework Programme for research, technological development and demonstration, under grant agreement number FP HEALTH 2013-602144.
Disclaimer This funding source had no role in the design of the study and will not have any role during the study execution, analyses, interpretation of the data or decision to submit results.
Competing interests None declared.
Ethics approval Regional Ethical Committee (CPP Ouest 1).
Provenance and peer review Not commissioned; externally peer reviewed.