Article Text

Abstract
Introduction This study protocol describes a trial designed to investigate whether high-flow heated and humidified nasal oxygen (HFHO) therapy in patients with hypercapnic acute respiratory failure (ARF) reduces the need of non-invasive ventilation (NIV).
Methods and analysis This is an open-label, superiority, international, parallel-group, multicentre randomised controlled two-arm trial, with an internal feasibility pilot phase. 242 patients with hypercapnic ARF requiring NIV admitted to an intensive care unit, an intermediate care or a respiratory care unit will be randomised in a 1:1 ratio to receive HFHO or standard oxygen in between NIV sessions. Randomisation will be centralised and stratified by centre and pH at admission (pH ≤7.25 or >7.25). The primary outcome will be the number of ventilator-free days (VFDs) and alive at day 28 postrandomisation. The secondary outcomes will encompass parameters related to the VFDs, comfort and tolerance variables, hospital length of stay and mortality. VFDs at 28 days postrandomisation will be compared between the two groups by Wilcoxon-Mann-Whitney two-sample rank-sum test in the intention-to-treat population. A sensitivity analysis will be conducted in the population of patients for whom the criteria of switching from NIV to spontaneous breathing, or conversely, are not strictly verified.
Ethics and dissemination The protocol has been approved by the Comité de Protection des Personnes (CPP) Sud-Ouest & Outre-Mer IV (ref CPP17-049a/2017-A01830-53) and will be carried out in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. A trial steering committee will oversee the progress of the study. Findings will be disseminated through national and international scientific conferences, and publication in peer-reviewed journals.
Trial registration number NCT03406572.
- acute respiratory failure
- non-invasive ventilation
- chronic obstructive pulmonary disease
- oxygenation
- hypercapnia
- tolerance
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- acute respiratory failure
- non-invasive ventilation
- chronic obstructive pulmonary disease
- oxygenation
- hypercapnia
- tolerance
Strengths and limitations of this study
First randomised trial to assess the effects of high-flow heated and humidified nasal oxygen therapy (HFHO) on ventilator-free days in patients with hypercapnic acute respiratory failure.
Multicentre, international study.
The criteria to switch between non-invasive ventilation and spontaneous breathing periods (HFHO or standard oxygen) obtained consensus among participating study investigators through a Delphi process.
The determination of the primary endpoint involves complex logistics, but the internal pilot phase will assess the feasibility of the full-scale study as currently designed.
Due to the nature of the intervention, blinding is not possible.
Introduction
Background and rationale
Chronic respiratory insufficiency and chronic obstructive pulmonary disease (COPD) are the third leading cause of death worldwide. With an estimated 1.5 million emergency room visits and 700 000 hospitalisations in the USA in 2010, they place substantial burdens on the patients, healthcare system and society.1 Patients decompensate at various stages of their disease and exhibit acute-on-chronic respiratory failure (ACRF), defined ‘as an inability of the respiratory pump, in concert with the lungs, to provide sufficient alveolar ventilation to maintain a normal arterial PCO2’ according to the British Thoracic Society.2 Hypercapnic acute respiratory failure (ARF) is a frequent cause of intensive care unit (ICU) hospitalisation.
Non-invasive ventilation (NIV) is the first-line ventilatory treatment for hypercapnic ARF. Its use has increased in hypercapnic ARF during the last two decades. Success of NIV increases with experience.3 4 It is applied intermittently or semicontinuously, that is, periods of spontaneous breathing (SB) with standard oxygen (O2) are interspersed between each NIV session.2 Depending on the severity of the respiratory failure and patient’s tolerance of the technique, length of NIV sessions may vary considerably. Figures on the total amount of NIV received during the first 24 hours vary between 7 and 20 hours. This means that patients may spend a noticeable amount of time with standard O2 alone during the acute phase of their respiratory failure. Standard O2 has several drawbacks that may limit the benefit of intermittent NIV in hypercapnic ARF: (1) a limited gas flow which is well below the patient’s inspiratory flow rate, (2) a limited capacity and efficiency of oxygenation with non-controlled inspired fraction of oxygen (FiO2), hence with a risk of excessive oxygen and induced hypercapnia and (3) cold and dry gas leading to discomfort and under-humidification of the airways and tracheobronchial secretions. Therefore, the benefits in terms of work of breathing and CO2 removal resulting from NIV sessions could be rapidly lost during the standard O2 periods.
Recently, the use of high-flow heated and humidified nasal oxygen therapy (HFHO) has gained enthusiasm among intensivists to manage ARF. HFHO delivers high flows (up to 70 L/min) of heated and humidified oxygen at a controlled and adjustable FiO2 (0.21–1) that rapidly improve respiratory distress symptoms, oxygenation, respiratory comfort and outcome of patients with hypoxemic ARF.5 Of note, all studies concur on the remarkable tolerance and comfort reported by patients treated with this technique.6 These unique features could overcome some of the drawbacks of standard O2 during SB periods in patients with hypercapnic ARF.
Aims and hypotheses
The primary aim of this study is to determine if the use of HFHO, as compared with standard O2, increases the number of ventilator-free days (VFDs) and alive at day 28, in patients with hypercapnic ARF requiring NIV. We hypothesise that HFHO applied during breaks of NIV could improve the management of ACRF in patients with chronic respiratory insufficiency and COPD, in comparison with standard O2. The use of HFHO could sustain the benefits of NIV, thereby reducing the amount of NIV required. The key secondary aims are to evaluate the effect of HFHO compared with standard O2 on:
Parameters related to VFDs.
Tolerance/comfort outcomes.
Hospital length of stay.
Mortality.
Side effects.
Methods and analysis
Design overview
The ‘High Flow-ACRF’ study is an open-label, superiority, international, parallel-group, multicentre randomised controlled trial (RCT), in which patients with hypercapnic ARF are allocated in a 1:1 ratio to HFHO (experimental group) or to standard O2 (control group). The trial design is summarised in table 1. We report the study protocol according to the SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) statement.7
Schedule of enrolment, interventions and assessments
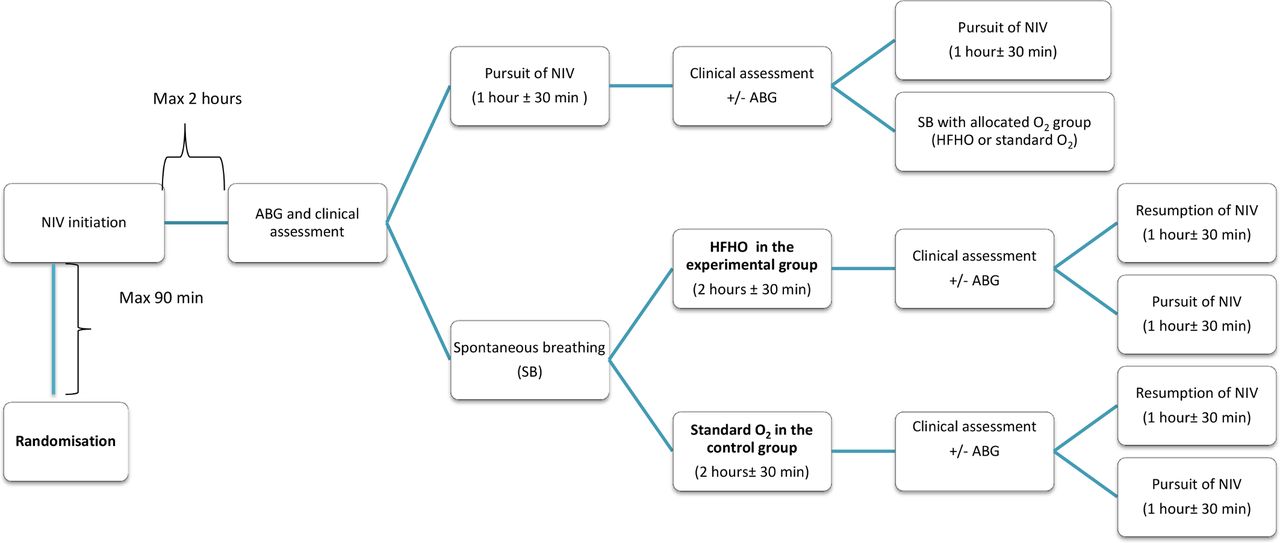
In both groups, treatment will start after randomisation with a first NIV session of 2 hours, with arterial blood gas (ABG) measurement between 1 and 2 hours after initiating the NIV session (a delay of 15 min following the end of NIV session in drawing the ABG sample will be permitted). The NIV will be extended for those patients with a pH <7.30. Patients will be assessed for their tolerance to NIV and their ability to switch to SB every 1 hour±30 min, except during sleep (22:00–8:00); they will be assessed for their tolerance of SB and for the need of resumption of NIV every 2 hours±30 min during the first 48 hours, and every 4 hours±1 hour thereafter.
To ensure that indications of NIV and invasive mechanical ventilation (IMV) are consistent across centres, NIV and IMV will be initiated and stopped in the same way in the two groups, using predefined criteria (box 1 and box 2). These criteria obtained consensus among participating study investigators, through a Delphi process (online appendix 1). Management of sequential NIV and oxygen therapy is described in online appendix 2. Patients will follow this cycle according to their allocated group, until weaning from NIV and subsequently from oxygen therapy (figure 1).
Criteria for switching between non-invasive ventilation (NIV) and spontaneous breathing (SB)
Switch from NIV to SB will be required if at least two criteria are met:
Improvement in clinical signs of moderate to severe respiratory distress, that is, one or more of the following criteria:
Respiratory rate ≤25/min.
Use of accessory respiratory muscles (from severe to moderate, or moderate to absent).
Paradoxical abdominal motion (yes/no).
Regression of signs of respiratory encephalopathy (sleepiness and/or asterixis and/or confusion) (yes/no).
Stabilisation or correction of hypercapnic respiratory acidosis on arterial blood gas (ABG), that is, pH ≥7.30 and decrease in PaCO2(alveolar carbon dioxide tension) of at least 10% as compared with baseline value (inclusion).
Stabilisation or correction of hypoxemia: 88% ≤SpO2 (oxygen saturation)≤92% and/or arterial oxygen tension (PaO2) ≥60 mm Hg (8 kPa) with FiO2 (inspired fraction of oxygen) ≤0.5 under NIV.
Intolerance to NIV (agitation and/or mask removal, and/or patient’s wish to interrupt session before).
Switch from spontaneous breathing to NIV will be required if one of the following criteria, at least, is met:
Worsening in clinical signs of moderate to severe respiratory distress, that is, one or more of the following criteria:
Dyspnoea (increase of at least two points in the Visual Analogue Scale).
Respiratory rate >25/min.
Use of accessory respiratory muscles (from absent to moderate, or moderate to severe).
Paradoxical abdominal motion (yes/no).
Signs of respiratory encephalopathy (sleepiness and/or asterixis and/or confusion) (yes/no).
No correction or impairment of hypercapnic respiratory acidosis on ABG, that is, one of the following criteria:
pH <7.30 and/or increase in PaCO2 of at least 20% as compared with value under NIV.
No correction or impairment of hypoxemia: SpO2 <90% and/or PaO2 <60 mm Hg (8 kPa) with standard O2≥5 L/min or FiO2 ≥0.5 with high flow heated and humidified nasal oxygen.
Predefined criteria for intubation (non-invasive ventilation (NIV) failure)
Patients will be intubated if, at least, one major or two minor criteria are met:
Major criteria
Respiratory or cardiac arrest.
Haemodynamic instability defined as a mean arterial blood pressure (ABP) ≤65 mm Hg despite fluid loading (≥500 mL) and/or the need of vasopressors.
Sustained cardiac arrhythmia.
Ventilatory inefficiency with agitation and/or major air leaks.
Minor criteria
Worsening of acute-on-chronic respiratory failure under NIV with:
Respiratory rate >35/min.
pH <7.20.
PaO2 (arterial oxygen tension) <60 mm Hg (8 kPa) with FiO2 (inspired fraction of oxygen) >0.6.
Neurological impairment (Glasgow Coma Scale <9).
Impossibility to clear copious tracheal secretions.
Organ failure other than respiratory.
Supplementary file 1
Supplementary file 2

{kind=link}
Ventilatory strategy for the first sequences of NIV and SB (HFHO or standard O2), according to respective predefined criteria. Patients will follow this cycle according to their allocated group, until weaning from NIV and subsequently from oxygen therapy. ABG, arterial blood gas; HFHO, high flow heated and humidified nasal oxygen; NIV, non-invasive ventilation; SB, spontaneous breathing.
Study setting and population
Participants will be prospectively recruited among patients admitted to an ICU, an intermediate care or a respiratory care unit, in 26 centres belonging to the European REVA network (24 centres in France, 1 centre in Spain and 1 centre in Switzerland). Patients will be considered eligible for randomisation if they fulfil all of the inclusion criteria and none of the exclusion criteria, as defined in box 3. The key eligibility criteria include hypercapnic ARF and the need for ventilation support.
Eligibility criteria
Inclusion criteria
Adult patients aged 18 or above, admitted to an intensive care unit (ICU), an intermediate care or a respiratory care unit.
Chronic respiratory failure known (documented) or strongly suspected on clinical, radiological and blood gases data and pulmonary function tests, in relation with an obstructive respiratory disease (chronic obstructive pulmonary disease (COPD), emphysema, overlap syndrome (COPD + obstructive sleep apnoea) or mixed (bronchiectasis, obesity hypoventilation syndrome)).
Patients requiring non-invasive ventilation (NIV) for hypercapnic acute respiratory failure (ARF) (whatever the precipitating cause), that is, with clinical signs of moderate to severe respiratory distress: dyspnoea and/or respiratory rate >25/min and/or use of accessory respiratory muscles and/or paradoxical abdominal motion and/or signs of respiratory encephalopathy (sleepiness, asterixis, confusion), and respiratory acidosis on arterial blood gases (defined by pH <7.35 and PaCO2 (alveolar carbon dioxide tension) >45 mm Hg despite the careful supply of oxygen and appropriate therapy that may include bronchodilators, corticosteroids and antibiotics).
Non-inclusion criteria
Contraindications to NIV.
Pure restrictive lung disease (thoracic deformity, neuromuscular pathology) and pure obstructive sleep apnoea.
Immediate need for intubation (respiratory or cardiac arrest).
Persistent haemodynamic instability (use of vasopressors for >1 hour).
Multiple organ failure (SOFA (sequential organ failure assessment) score >6).
NIV treatment for >3 hours before admission to the ICU, intermediate care or respiratory care unit.
Anticipated difficulties to conduct NIV (facial trauma or deformation, edentulous patient).
End-stage chronic respiratory insufficiency (defined as the use of NIV at home).
Non-treated pneumothorax.
Impossibility to perform subjective assessment of dyspnoea and comfort (cognitive impairment).
Patient under guardianship or trusteeship.
Pregnancy/breast feeding.
Current participation in another clinical trial with an endpoint related to NIV.
No affiliation to social security (beneficial or assignee).
Lack of oral informed consent (express consent) from the patient or relative of appropriate. For those patients who are unable to give written informed consent at the time of enrolment due to the severity of their illness, a process of delayed consent will be used.
Interventions
Experimental group
In-between each NIV session, oxygen will be delivered using HFHO (AIRVO 2; Fisher & Paykel Healthcare, Auckland, New Zealand) with a flow set between 50 and 60 L/min, and a FiO2 set to reach a targeted oxygen saturation (SpO2) of 90% ≤ SpO2 ≤94%.
Control group
NIV will be initiated based on the same criteria and with the same parameters as the HFHO group. In-between each NIV session, oxygen will be delivered using low flow of standard O2 to reach the same targeted SpO2 of 90% ≤ SpO2 ≤94%.
Trial outcomes
Primary outcome
The primary outcome is the number of VFDs during the 28 days after randomisation.
It is defined as the number of days between randomisation and day 28 where the patient is alive and breathes without any ventilatory support (NIV or IMV) for at least 48 consecutive hours.8 Since the duration of follow-up of intervention trials addressing lung function in the critically ill is typically 28 days after study enrolment, and most of patients with ARF have either died or successfully weaned from ventilation by day 28, the 28 day landmark is chosen to determine VFDs.
Secondary outcomes
Please see table 2 for the full list of secondary outcomes.
Secondary outcome measures
Randomisation and sequence generation
The randomisation will be performed using CleanWEB, which is an online, central randomisation service running 24 hours/24. The randomisation groups will be indicated by the server and confirmed by email. In case of dysfunction of the server, the allocated randomisation group will be provided by phone by the coordinating office (Unité de Recherche Clinique, Bichat Hospital) and confirmed by email. The patient randomised by this method must be registered thereafter on CleanWEB server, before the next randomisation. The randomisation sequence will be computer generated in advance by a statistician of the coordinating office. It will be stratified by centres and pH at admission (pH≤7.25 or pH>7.25), with variable block sizes.
Allocation concealment
The number of experimental units per block will be kept confidential to avoid prediction of future patient’s allocation. Only the independent statistician and the computer programmer who will implement the sequence assignment in the secure electronic case report form (eCRF) will have access to the randomisation list. Allocation concealment will be ensured, as CleanWeb services will not release the randomisation code until the patient has been recruited into the trial.
Blinding
Due to the nature of the intervention, it is not feasible to blind study participants and investigators to whether the participants are allocated to the HFHO or O2 group. The study statistician, however, will be blinded to the groups.
Protocol feasibility
We will undertake a preliminary pilot study in five centres before conducting the full-scale research project. This preparatory phase will last 5 months (including an enrolment period of about 3 months) and seek to recruit approximately 24 patients. We aim to address current uncertainties regarding the feasibility of performing the assessments for switching between NIV and SB according to the study protocol. The outcomes to assess feasibility are described in box 4. The key criterion for evaluating the success of the pilot study is that at least 90% of patients will have valid and complete VFDs data. Failure to reach these criteria does not necessarily indicate unfeasibility; rather, it underlines modifications to be made in the protocol. Data from the pilot phase will be included into the final analysis if no modification has been made in the assessment of the VFDs.9
Outcomes of the pilot study to assess feasibility
Missing data rate for the primary outcome measure of the main trial (number of patients with missing ventilator-free days).
Duration (hours) of non-invasive ventilation (NIV) sessions and invasive mechanical ventilation.
Proportion of patients achieving 48 consecutive hours of unassisted breathing.
Mortality rate
All-cause mortality at day 28 postrandomisation.
All cause in-hospital mortality.
All cause inpatient mortality in the unit where the patient has been first admitted (either intensive care unit (ICU), intermediate care or respiratory care unit).
All cause inpatient mortality in the unit where the patient has been transferred to further receive NIV (eg, from ICU to intermediate care, for those who provide NIV in intermediate care).
All cause inpatient mortality in the units where the patient has received NIV.
Proportion of patients with timing of the assessments respected according to the protocol of the main study (see ‘Procedure for the research’ section in the main protocol).
Eligibility rate and reasons for exclusion (number of patients meeting the inclusion criteria divided by the number of patients with suspected or known chronic respiratory insufficiency, admitted to an ICU, an intermediate care or a respiratory care unit).
Consent rate (number of patients who consented to participate divided by the number of patients eligible for the study), and reasons for not participating in the study.
Retention rate (number of patients who remained in the study).
Statistical considerations
Sample size calculation
The most recent large study dataset on hypercapnic respiratory failure patients comes from the EchoICU trial.10 In that study, mean duration of NIV was 5.2 days. Fifteen per cent of patients failed NIV and required intubation. A total sample size of 242 patients (121 in each group) was determined necessary to detect as statistically significant a decrease of 40% of ventilator support duration in the HFHO group, with a power of 90% and an alpha risk of 0.05. The sample size calculation was based on the following assumptions: mean number of VFDs between day 0 and day 28 of 21.3 in the standard O2 group, which is congruent with EchoICU trial data8; mortality rate at day 28 of 10%11 and equal in both groups; a loss to follow-up rate of approximately 10%.
Statistical analysis
Data will be analysed using SAS software (V.9.4 or higher). A flow chart will describe the number of eligible patients, the number of patients effectively included in the study and in each of the two groups. Demographic and clinical characteristics of the study patients at baseline and at each follow-up visit will be reported with frequencies and percentages for categorical variables, and with mean, SD, median and IQR for continuous variables. This description will be performed for all patients and for each group. All analysis for the primary and secondary outcomes will be performed in the intention-to-treat population. All tests will be two-sided with a statistical significance of 5%.
Primary outcome analysis
VFDs at 28 days postrandomisation will be compared between the two groups by Wilcoxon-Mann-Whitney two-sample rank-sum test in the intention-to-treat population. A sensitivity analysis will be conducted in the patient population for which the criteria for switching from NIV to SB, or vice versa, are not strictly verified. Imputation methods will be used to replace missing values from the primary outcome. The MICE method ‘Multivariate equation imputation by chain equations’ will be used.
Secondary outcome analyses
Continuous outcomes (including length of hospital stay, duration to reach predefined criteria to stop NIV, duration of NIV and IMV, patient comfort, respiratory rate and dyspnoea assessment score) will be compared between the study groups using Student’s t-test or the Wilcoxon-Mann-Whitney two-sample rank-sum test as appropriate. Dichotomous outcomes (including mortality, intubation, complications related to NIV, complications related to standard O2 and HFHO, premature NIV cessation and use of accessory muscles) will be compared using χ2 or Fisher’s exact tests as appropriate. Intubation-free survival (time to intubation) will be calculated from enrolment to time to intubation. Comparison of time to intubation between the two groups will be performed using the Kaplan-Meier survival function with a log-rank test to assess significance. Data will be censored at discharge from the unit where the patient receives assisted ventilation (whether it is the unit of admittance or the unit where the patient has been transferred to). No interim analysis is planned.
Subgroup analyses
A subgroup analysis is planned to analyse whether the treatment effect is modified by pH before randomisation. Another subgroup analysis will investigate whether the treatment effect is modified by a history of NIV use before inclusion into this study. Both analyses will be conducted for the primary outcome (VFDs).
Patient and public involvement
The research question was developed thanks to our patients’ input and experience with NIV and nasal high flow. Patients are regularly questioned on the benefits of these techniques regarding comfort and tolerance. Patients’ constant feedback during routine care therefore helped us assess the burden of the intervention and establish the study design. For example, our initial design of the study included evaluation of patients’ respiratory parameters and interruption of NIV during night time. Based on the patients’ feedback—that it would be best for their own comfort, not to be woken up for the assessment—we modified the study protocol, so as to preserve their sleep. Results of the trial will be made available to all participants via ClinicalTrials.gov as well as by email notification.
Trial status
Recruiting.
Ethics and dissemination
Legal obligations and approval
Sponsorship has been agreed by Assistance Publique—Hôpitaux de Paris (AP-HP, Clinical Research and Innovation Department) for this minimal risks and constraints human research study. AP-HP has obtained the favourable opinion of the Comité de Protection des Personnes (CPP) Sud-Ouest & Outre-Mer IV (ref CPP17-049a/2017-A01830-53) for the study protocol (version HIGH-FLOW ACRF −01; 17 October 2017). The AP-HP has sent the CPP approval and the summary of the protocol to the Agence Nationale de Sécurité du Médicament et des Produits de Santé for information. The trial will be carried out in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines. Any substantial modification to the protocol must be sent to the sponsor for approval. Once approval has been received from the sponsor, it must also obtain approval from the CPP before the amendment can be implemented. The information sheet and the consent form can be revised if necessary, particularly if there is a substantial amendment to the study or if adverse reactions occur. AP-HP is the owner of the data. The data cannot be used or disclosed to a third party without its prior permission.
Methods for obtaining information and consent from research participants
In accordance with Article L.1122-1-1 of the French Public Health Code, no minimal risk and constraints research can be carried out on a person without his/her free and informed consent, obtained expressly after the person has been given the information specified in Article L.1122-1 of said Code. For both organisational and practical reasons, we will seek oral rather than written consent. The person will be given 15 min between receiving the information and being asked to give their consent. The person’s free and informed oral consent will be obtained by the principal investigator or a physician representing the investigator, a maximum 1 hour after being admitted to the service.
Emergency inclusions will be allowed in this research protocol, in accordance to the Public Health Code, article L1122-1-2. The patient and legal representative will be informed as soon as possible, and a written informed consent will be required for the pursuit of this research. The patient and legal representative will also have the right to oppose to the use of his data in the research. For patients who are not able to sign the informed consent form, the consent may be obtained, in descending order of priority, from a legal representative, family member or a close relative, if they are present. The investigator will specify in the research participant’s medical file the methods used for obtaining their consent, as well as the methods used for providing information to obtain consent. The investigator will retain the original signed and dated consent form.
Data collection and quality control
The persons responsible for the quality control of clinical matters will take all necessary precautions to ensure the confidentiality of information relating to the study participants. These persons, as well as the investigators themselves, are bound by professional secrecy. During or after the research, all data collected about the participants and sent to the sponsor by the investigators (or any other specialised collaborators) will be anonymised. Under no circumstances should the names and addresses of the subjects involved be shown.
In any case of premature withdrawals and exits, the investigator must document their reason(s) and try to collect primary endpoint, secondary endpoints and safety assessment, if the participant agrees. If a participant exits the study prematurely or withdraws consent, any data collected prior to the date of premature exit may still be used, excepted if the participant refuse in writing.
A data monitoring committee (DMC) is not required for this research with minimal risks for the following reasons: (1) the devices have been used in routine for many years, and so we have enough perspective regarding their safety; (2) none of the three international experts who reviewed the protocol during the funding application process stressed the need for a DMC and (3) according to the French law, research with ‘minimal risks’ does not require a DMC. The research data will be collected and monitored using an eCRF through CleanWEB Electronic Observation Book and will be centralised on a server hosted by the AP-HP Operations Department. This research is governed by the CNIL ‘Reference Method for processing personal data for clinical studies’ (MR-001, amended). AP-HP, the sponsor, has signed a declaration of compliance with this ‘Reference Method’.
Research staff of the URC will work with local investigators to obtain data that are as complete and accurate as possible. An independent Clinical Research Associate appointed by the sponsor will be responsible for the proper running of the study, for collecting, documenting, recording and reporting all handwritten data, in accordance with the Standard Operating Procedures applied within the Clinical Research and Innovation Department of AP-HP. The investigators agree to accept the quality assurance audits carried out by the sponsor as well as the inspections carried out by the competent authorities. All data, documents and reports may be subject to regulatory audits. These audits and inspections cannot be refused on the grounds of medical secrecy. An audit can be carried out at any time by independent individuals appointed by the sponsor. The aims of the audits are to ensure the quality of the study, the validity of the results and compliance with the legislation and regulations in force. The persons who manage and monitor the study agree to comply with the sponsor’s audit requirements. The audit may encompass all stages of the study, from the development of the protocol to the publication of the results and the storage of the data used or produced as part of the study. Sponsor is responsible for access to the study database.
Safety considerations
During this research, adverse events (serious (SAE) and non-serious) do not need to be reported to the sponsor. The report must instead be made as part of the vigilance procedure applicable to the intervention under investigation. However, the investigator will report SAE and non-serious adverse event in the ‘Adverse events’ section of the e-CRF. The following events are foreseen, expected SAEs:
SAE associated with the use of NIV:
Pneumothorax.
Stopping NIV for intolerance/agitation leading to intubation.
SAE potentially associated with the use of HFHO:
Pneumothorax (this is only theoretical, as it has never been described).
SAEs that are not specifically linked to the experimental protocol but are the result of the progression of the disease:
Worsening of hypoxemia or worsening of hypercapnia requiring intubation and IMV.
Cardiovascular event (cardiac arrest, myocardial ischaemia, supraventricular or ventricular abnormal cardiac rhythm).
Initiation of IMV (except for those situations where the investigator believes there is a link between the SAE and the experimental protocol).
Nosocomial pneumonia, ventilator-associated pneumonia.
Occurrence of organ failure or septic shock.
Death related to the progression of the disease that led the patient to the ICU, intermediate care or respiratory care unit.
Death related to the progression of a chronic disease mentioned in the patient’s medical history.
Death resulting from a decision of withholding or withdrawing life support.
Dissemination of results
Findings will be disseminated through national and international scientific conferences, and publication in peer-reviewed journals. The results of the trial will be relevant to intensivists who manage patients with hypercapnic ARF.
Discussion
To date, NIV is the cornerstone of hypercapnic ARF ventilatory support, and standard O2 is used in-between breaks of NIV. Because of several drawbacks regarding its characteristics (absence of positive end-expiratory pressure, cold and not humidified, and limited flow), standard O2 may not be the best delivery mode to maintain the beneficial effects obtained during NIV periods. HFHO has shown its unambiguous benefits during hypoxemic respiratory failure; many of its features may apply to hypercapnic respiratory failure.
This trial is—to the best of our knowledge—the first RCT that will assess the use of HFHO in patients with hypercapnic ARF. To date, outside case reports in patients with hypercapnic ARF,12 the only data available on HFHO in this indication has been obtained from small series of stable patients with COPD.13–16 These observational studies have consistently shown that HFHO is feasible, well accepted and exerts beneficial effects in patients with severe but stable COPD. Improvements have been measured in respiratory rate, tidal volume, rapid shallow breathing index, PaCO2 and end-expiratory lung volume, all suggesting that HFHO enables a certain decrease in the work of breathing in these patients.
A randomised cross-over trial has confirmed improvement in respiratory parameters in stable patients with COPD.17 In the acute setting of hypoxemic respiratory failure, one study has shown that HFHO was better tolerated than NIV when used sequentially.18 When specifically addressing the breaks in-between NIV sessions, one single RCT has evaluated HFHO in the setting of ARF.19 It planned to enrol 70 patients with ARF requiring NIV. Patients were randomised to receive either standard O2 or HFHO in-between NIV sessions. In this study, the primary endpoint was the time spent without NIV. Secondary endpoints included respiratory variables, a dyspnoea score and patients’ comfort. To date, preliminary results have been published under abstract form, for the first 44 patients. No difference was found for the primary outcome. Similarly, respiratory variables did not differ. However, dyspnoea and comfort were significantly improved with the use of HFHO. There are, however, several limitations that need to be acknowledged: (1) the number of patients is relatively small; (2) the study was not limited to hypercapnic patients (only a small number of patients with COPD among the first 44 patients included) and (3) there was significantly more patients with hypoxemic ARF in those allocated to HFHO; these patients may have required longer NIV treatment. In the context of hypoxemic ARF, a recent retrospective study showed that patients with chronic lung disease and treated with HFHO had similar outcomes compared with those without a history of chronic lung disease.20 Taken together, these data suggest that patients with hypercapnic ARF could also benefit from this technique.
Strengths and limitations
This is the largest prospective multicentre RCT comparing standard O2 to HFHO in the field of hypercapnic ARF. Very strict criteria to initiate, stop and resume NIV will be used to ensure that all centres will apply the technique similarly. A consensus on these criteria has been reached through a two-round Delphi survey. A pilot study will assess the feasibility of these criteria for the trial. Regarding the blinding, because of the nature of the study and the devices evaluated, it is not possible to blind patients nor the investigators to the allocation group. However, all the analyses will be carried out in a blinded manner.
In conclusion, the ‘High Flow-ACRF’ trial is an investigator-initiated RCT empowered to test the hypothesis that HFHO in comparison to standard O2 in-between breaks of NIV sequences may increase the number of VFDs in patients with hypercapnic ARF. Its results could be very useful for optimising ACRF management with NIV in clinical practice.
References
Footnotes
Contributors Trial design development: J-DR, CG, FD and JM. Design of the statistical analysis plan: FD and ME-F. Drafting of the present manuscript: FD and J-DR. All authors have contributed and approved this manuscript.
Funding This work was supported by French Ministry of Health (Programme de Recherche Clinique 2016 (PHRC), project no: PHRCN-16-0383). Fisher & Paykel will supply Airvo2 devices, necessary equipments and consumables and covered the costs of the two foreign centres not covered by the PHRC.
Competing interests J-DR, JM and CG had travel and accommodation expenses to attend scientific meetings covered by Fisher & Paykel.
Patient consent Obtained.
Ethics approval Comité de Protection des Personnes (CPP) Sud-Ouest & Outre-Mer IV.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement We envisage no additional data that would not be published.
Collaborators From the REVA network: Stephan Ehrmann (Tours), Alain Mercat (Angers), Alexandre Demoule (Paris), Muriel Fartoukh (Paris), Benjamin Sztrymf (Clamart), Jack Richecoeur (Beauvais), Claude Guerin(Lyon), Jean Reignier (Nantes), Saad Nseir (Lilles), Arnaud Thille (Poitiers), ErwanL ’Her (Brest), Thierry Boulain (Orléans), Jean-Philippe Rigaud (Dieppe), AntoineCuvelier (Rouen), Kaldoun Kuteifan (Mulhouse), Sandrine Jaffré (Nantes), Armand Mekontso Dessap (Créteil), Jean-Christophe Callahan (Le Mans), Jean-Claude Lacherade(La Roche sur Yon), Jean-Michel Constantin (Clermont-Ferrand), Gilles Capellier(Besançon), Emmanuel Antok (St-Pierre), Sarah Heili (Madrid) ; Lise Piquilloud (Lauzanne)