Article Text

Abstract
Introduction 0.9% saline and Ringer’s lactate are the two most common resuscitation crystalloid fluids. 0.9% saline may lead to hyperchloraemic metabolic acidosis and may be associated with impaired kidney function and death. Few large multicentre randomised trials have been conducted to evaluate the effect of these two fluids on clinically important outcomes.
Methods FLUID is a pragmatic pilot cluster randomised crossover trial in which four hospitals will be randomised to normal saline or Ringer’s lactate for 14 weeks, then crossover to the alternative fluid for the subsequent 14 weeks after 1 to 3 week transition. With waiver of informed consent, all adult and paediatric patients admitted to participating sites will be included in the FLUID trial except for neonates. Primary feasibility outcome is study fluid protocol adherence (target:≥80%). Secondary feasibility outcomes include time to research ethics board (REB) approval and readiness to trial initiation (≤3 months from REB submission and approval). Primary (composite of death or re-admission to hospital in first 90 days of index hospitalisation) and secondary clinical outcomes for the future large FLUID trial will be described. Protocol adherence will be collected by site at specified time points. All clinical data will be obtained at patient level through provincial health administrative data held at the Institute for Clinical Evaluative Sciences (ICES). Event rates for the primary and secondary outcomes will be described using frequencies and proportions with 95% CIs. Intracluster and interperiod correlation coefficients will be calculated from population-level data available at ICES.
Ethics and dissemination The study protocol has been approved by the Ottawa Health Science Research Ethics Board. The FLUID pilot will determine feasibility, and ICES data across all potential sites in Ontario will allow calculation of sample size parameter estimates to inform the design and implementation of the large trial.
Trial registration number NCT02721485; Pre-results.
- crystalloid fluid
- ringer’s lactate
- cluster cross over rct
- fluid therapy
- 0.9% Saline
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Pilot trial to inform the rigorous design and conduct of a large-scale cluster randomised trial addressing a question of high importance to patients and the healthcare system.
Highly pragmatic because the trial will evaluate the effect of crystalloid resuscitation fluid within the context of the usual care delivered to these patients on a daily basis.
It is a cost-efficient trial design using innovative methods to capture all relevant outcomes from provincial health administrative data sources without the need for patient recruitment.
Integral involvement of ethicists on the FLUID team to ensure that the highest ethical standards and patient protections are maintained.
Inability to collect fluid protocol exposure and adherence for individual patients and the small number of clusters limits external validity.
Introduction
Other than the administration of oxygen, crystalloid resuscitation fluids including 0.9% saline and Ringer’s lactate are among the most common interventions administered to hospitalised patients.1 2 These fluids may be used as a life-saving measure to re-establish haemodynamic stability, for rehydration, and to replace fluid losses and maintain intravascular volume in the surgical setting. Saline 0.9% contains more sodium and more chloride in comparison to Ringer’s lactate.3 In addition to containing small amounts of potassium and calcium, Ringer’s lactate contains lactate as a buffer and is relatively hypotonic in comparison with 0.9% saline4 (see the online supplementary file 1).
Supplementary file 1
Varying levels of evidence have led to question the safety of 0.9% saline due to its high chloride content and its association with the development of hyperchloraemic metabolic acidosis.5–7 Ringer’s lactate is considered a balanced crystalloid8 9; it contains a chloride concentrations that is closer to plasma, and in combination with its lactate buffer that gets converted to bicarbonate in the liver, does not induce acidosis.1 Balanced crystalloids are more expensive than 0.9% saline (approximate cost of 1 L bag of 0.9% saline $1.30 and Ringer’s lactate $1.66 at the Ottawa Hospital, Ontario, Canada) and they are not without the potential for adverse effects. For example, Ringer’s lactate contains a buffer which can cause metabolic alkalosis10 11 and theoretically, has the potential for arrhythmias, tetany, coma and seizures.12–14 Although the lactate in Ringer’s lactate does not cause a metabolic acidosis, it may accumulate in the setting of liver failure and may influence clinical diagnoses and clinical decision making.15–17 Moreover, Ringer’s has a lower osmolarity in comparison with 0.9% saline and when administered rapidly in large volumes could theoretically reduce plasma osmolarity and increase the risk of oedema formation,18 which may be concerning for patients with cerebral oedema.
Studies in healthy volunteers, the critically ill and surgical populations have associated 0.9% saline with an increased risk of acute renal injury and requirement for dialysis,19–23 postoperative infections,23 death,23 24 and increased resource and blood transfusion use.23 However, many of the studies suffer from methodological weaknesses including confounding by indication, selection bias and inability to ascertain the effects of a specific fluid due to co-interventions administered. Two systematic reviews of randomised trials have examined the effects of balanced (low chloride) crystalloid fluids versus 0.9% saline. One Cochrane review included 13 trials and 706 patients of heterogeneous surgical populations (range of sample sizes of individual trials: 24–120).5 The other review included a total of 28 trials and 1368 patients (range of sample sizes of individual trials: 20–108).6 None of the individual trials included in these reviews were powered for clinically important outcomes such as death or re-admissions to hospital. They focused on physiological outcomes such as acid–base measures, electrolytes, pH, lactate and creatinine levels. Another systematic review that focused on resuscitation fluids in sepsis included an analysis of indirect comparisons of 0.9% saline with balanced crystalloids and found non-significant reductions in death (OR 0.78, 95% CI 0.58 to 1.05)25 and the requirement for renal replacement therapy (OR 0.85, 95% CI 0.56 to 1.30).26 A systematic review of 15 randomised trials published in 2018 compared low chloride to high chloride fluids in critically ill (6 trials, n=3468) and perioperative adult patients (9 trials, n=583 patients) with primary outcomes of death and requirement for renal replacement therapy.7 In the review, there were no significant differences in death (OR 0.90, 95% CI 0.69 to 1.17) or requirement for renal replacement therapy (OR 1.12, 95% CI 0.80 to 1.58) for the low chloride as compared with high chloride fluid groups. Authors rated the quality of evidence as low but also imprecise with insufficient power to detect small but clinically important differences between the fluids. Two pilot cluster randomised controlled trials in the intensive care unit (ICU) that were included in this review compared 0.9% saline with either Ringer’s lactate or Plasma-lyte which is another balanced crystalloid fluid.27 28 Neither of these trials detected differences in clinical outcomes between the fluid groups although they were not powered adequately to do so. Furthermore, two single institution multiple monthly crossover studies comparing 0.9% saline with balanced crystalloids (Ringer’s Lactate and Plasma-Lyte) conducted in the emergency department (SALT-ED)2 and ICU (SMART)29 found small differences in the ‘Major Adverse Kidney Events within 30 days’ composite outcome which includes death, requirement for dialysis or persistent renal dysfunction in favour of balanced crystalloids. Authors and editorialists assert that it is essential to conduct large robust multicentre randomised trials5 24 30–34 comparing 0.9% saline with balanced crystalloid fluids. Furthermore, Myburgh’s editorial on the SMART trial of critically ill patients suggests that longer term, clinically relevant and patient-centred outcomes that are supported by health economic evaluations are required to provide confirmatory evidence to guide future usual care crystalloid resuscitation fluid clinical practice and related healthcare resource allocation.34 We agree with this point and would extend it to include the lens of the entire hospital since these fluid interventions are so widely applied.
The large FLUID trial will examine whether Ringer’s lactate reduces the incidence of death and hospital re-admissions compared with 0.9% saline in all patients admitted to hospital. Prior to embarking on our large-scale trial, we will undertake the FLUID pilot trial to examine feasibility related to study fluid protocol adherence, time to research ethics board (REB) approvals and time to readiness to initiate the trial.
Methods and analysis
Study design
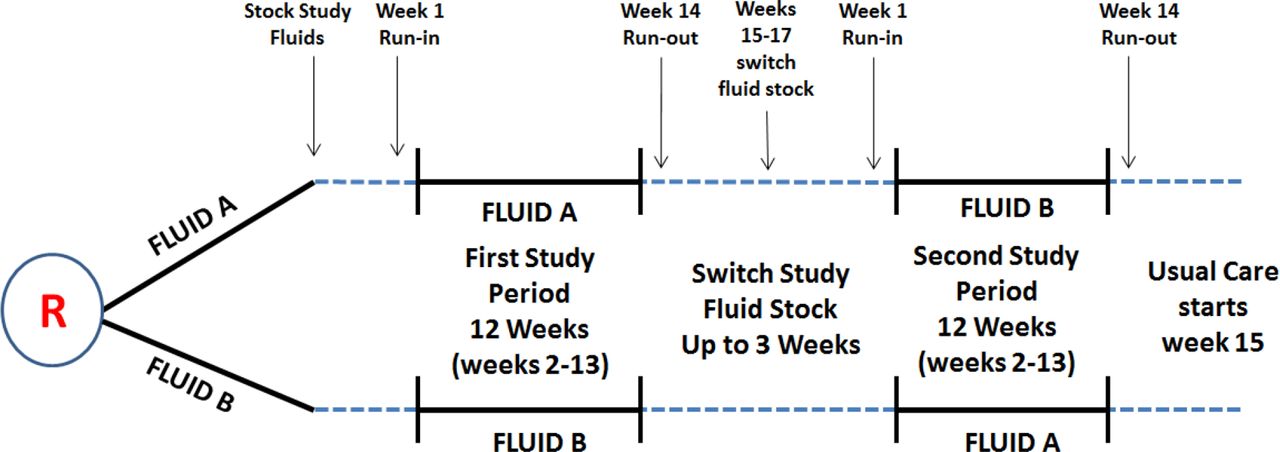
The FLUID pilot trial is an open-label hospital wide cluster crossover pragmatic randomised trial (see figure 1). A cluster randomised design was chosen for several reasons. First, it is essential to have the same study fluid available throughout the hospital to minimise contamination and maximise adherence to the study fluid to generate unbiased estimates. Second, from the perspective of trial logistics and cost, it would be extremely challenging to randomise enough individual patients from all areas in the hospital in a large trial. Third, the availability of health administrative data for all hospitals in the province of Ontario, Canada creates a unique opportunity to conduct a large-scale, cost-effective pragmatic trial without the need to recruit individual patients for data collection. The crossover design was selected as it improves the power and statistical efficiency of the trial, and minimises confounding because each cluster will essentially serve as its own control group.

{kind=link}
Study fluid for the first study period will be stocked from 1 to 3 weeks before initiation of the 1-week run-in period. No patients admitted during the 1-week run in-period will be included in the analysis. The 1-week run-in period will familiarise hospital staff (physicians, nurses and trainees) with the FLUID operations, including the FLUID automatic substitution orders, prior to initiation of the active 12-week study fluid period (weeks 2–13), where all patients with index hospitalisations are included. To ensure patients admitted in week 13 receive the same study fluid, a run-out period (week 14) will be instilled and the same fluid from the study period will remain stocked on the shelves throughout the hospital. No patients admitted during the 1-week run-out period will be included in the analysis. We will allow hospitals up to 3 weeks (weeks 15–17) to swap out the study fluid and crossover to the other study period fluid before the second 1-week run-in period begins.
Study setting
Four hospitals will participate in the FLUID pilot trial. Three hospitals are tertiary care centres and the fourth is a community hospital. Representation of academic and community hospitals will allow the identification of feasibility challenges that will then be addressed in preparation for the future large FLUID trial. For pilot site selection, we have recruited a convenience sample of hospitals where the Principal Author practices and through the Canadian Critical Care Trials Group (because FLUID is part of this network) and the direct contact of a critical care physician at a community centre located in the same city as the FLUID co-ordinating centre in Ottawa, Ontario.
Objectives
Primary objectives
The primary objective of this pilot trial is to examine the feasibility of conducting a larger pragmatic comparative effectiveness cluster crossover trial in hospitalised patients. The larger trial will examine the effect of fluid therapy with Ringer’s lactate versus 0.9% saline on the composite outcome of mortality or the requirement for re-admission to hospital within the first 90 days of the index hospital admission. Our feasibility objectives for the pilot trial are to evaluate study fluid protocol compliance (target:≥80%), time to REB approvals at each site (target:≤3 months (90 days) from REB submission) and time to readiness to initiate the clinical trial (target:≤3 months (90 days) from REB approval). Using these prespecified success criteria, we hypothesise that a large and adequately powered cluster randomised trial will be feasible.
Secondary objectives
Our secondary objectives for this pilot trial are to assess the feasibility of using health administrative data housed at a Canadian provincial health administrative database (Institute for Clinical Evaluative Sciences) in Ontario to measure and quantify our primary (death and hospital re-admissions) and secondary outcomes (requirement for dialysis, need for re-operation, need for re-intubation postoperatively, length of hospital stay, emergency department visits and discharge to a facility other than home).
Eligibility criteria
Inclusion criteria
Hospital level: all participating hospitals must have a level II or III ICU because these hospitals have the capability of admitting patients that are more severely ill and that may require critical care support for one, or two or more failed organs, respectively (eg, mechanical ventilation and dialysis)35; more severely ill patients tend to receive greater amounts of crystalloid fluid which may increase the ability to detect a treatment effect. Patient level: all adult and paediatric patients admitted to the participating hospitals for the first time in the last 90 days (index admission) over the duration of the study period will be included in FLUID. These include patients who are admitted through the emergency department, direct admissions to the hospital ward, and patients who are admitted to hospital and undergoing surgery. Although paediatric patients are not excluded from FLUID, only paediatric patients who are admitted through the same emergency room as adults in our participating hospitals (more common from community hospitals) will be included as we are not recruiting paediatric specialty hospitals to participate in FLUID. Because true fluid exposure status is not recorded in administrative databases, all admitted patients will be included, regardless of actual fluid administration. This approach is reasonable as the majority of patients receive either 0.9% saline or Ringer’s lactate as fluid boluses or as infusions for the treatment of dehydration, haemodynamic instability and for the replacement of fluid losses during their hospital admission.
Exclusion criteria
Hospital level: we will exclude hospitals that have fewer than 6000 acute care admissions per year (<1500 admissions per study period). Although the exclusion of smaller hospitals may limit the generalisability of our study results, it will help to reduce excessive between-hospital variability in our outcome estimates and cluster sizes, thus improving the trial efficiency and reducing the number of sites required to detect a treatment effect. Patient level: the only patient population that will be excluded in FLUID are neonates since Ringer’s lactate is neither used nor recommended for use in this population.36 Although there are no other overt exclusion criteria, physicians may also opt out of the use of the allocated study fluid for a specific patient if they believe they have a strong clinical reason to do so (eg, severe brain injury with raised intracranial pressure). Such patients will nevertheless be included in analyses as exposure status will not be recorded in the trial.
Randomisation, allocation concealment and masking
In the pilot trial, sites will be randomised sequentially after institutional contract agreements have been signed and REB approvals have been obtained. Figure 1 provides a summary of the FLUID trial design. Two of the hospitals will be allocated to begin the trial with 0.9% saline as the control, while the other two will be allocated to Ringer’s lactate as the treatment. FLUID has two 14-week study periods. For each study period, week 1 will serve as a run-in, weeks 2 –13 as the study period time during which time all patients with index admissions to the study hospital will be included for analysis. Week 14 will serve as a run-out week during which time the study fluid remains stocked in the hospital for use by patients admitted during weeks 2–13; however, new patients admitted during week 14 will not contribute to the analysis. After the 1-week run-out period, hospitals will have up to an additional 3 weeks to crossover to period two study fluid.
The allocation of hospitals to begin with 0.9% saline versus Ringer’s lactate will be according to computer-generated random numbers at the coordinating centre prepared by a statistician not familiar with the sites. Only the statistician will have access to the allocation sequence which will be concealed from study investigators and sites until sites are ready to initiate the trial. FLUID will be an open-label clinical trial since the cost of blinding the fluids throughout each hospital is logistically and financially prohibitive. However, the risk of reporting bias is minimal as our clinical outcomes for the pilot trial and future large trial are objective and will be obtained using provincial health administrative data (housed at Institute for Clinical Evaluative Sciences) in Ontario.
Trial preparation and conduct strategies
Trial preparation
Several strategies will be implemented in advance of trial initiation to optimise site ‘buy in’ and FLUID education. These include: (1) trial approval by the Medical Affairs Committee (MAC) and the Head of Nursing at each of the participating hospitals; (2) trial approval by REBs in Ontario; and (3) implementation of an education campaign which includes information about MAC, head of nursing, and REB approval in advance of trial initiation for the physicians and nurses in the participating hospitals. To develop a FLUID educational campaign that is tailored to each site, the principal investigator (LM) and FLUID study manager (TM) will communicate with key stakeholder groups (eg, site principal investigator, head of nursing, head of medical affairs and head of purchasing/inventory control) at each site to discuss the protocol and to identify potential site-specific barriers to, and facilitators of, trial implementation. With this information, the FLUID study manager, site research coordinator and site principal investigator will implement a multimodal communication strategy to educate physicians and trainees, nurse educators, managers and bedside nurses, and inventory personnel about the FLUID trial. Communication strategies will include face-to-face discussions about the FLUID trial rationale, design and study outcomes at department and staff meetings, nursing educational forums and during nurse huddles, and written communication about FLUID with use of standardised emails, posters, newsletters and a FLUID icon that will be available on clinical computer desktops throughout the hospitals (see table 1 for strategies as well as the timing for roll-out in relation to study start-up for each strategy). Some strategies will be targeted to specific stakeholder groups. For example, nurses will be reminded to not co-administer Ringer’s lactate with ceftriaxone or blood products as there is a risk of salt precipitation with the former, and a potential risk for clotting with the latter due to calcium in Ringer’s lactate. Provision of a key fact sheet for nursing and a quick guide of Y-site compatibility with Ringer’s lactate and commonly prescribed medication will also be distributed to nurses via nurse educators and/or clinical managers.
FLUID communication strategies before and during the conduct of the study
Trial conduct
In the week before FLUID is initiated, stakeholder group leads will confirm readiness to start the trial with the FLUID study manager; inventory personnel will also confirm that they have adequate inventory of both study fluids for the two study periods in advance of trial initiation. During the conduct of FLUID, the following strategies will be implemented to maintain staff awareness of FLUID and to maximise compliance to the study fluid during each study period: (1) during the first week of study period 1 and 2, the FLUID site research coordinator will visit the wards and units to ensure the appropriate amount of study fluid is available and bright signage is in place where the fluids are stocked to ensure that the nurses are reminded to autosubstitute the study fluid; (2) the FLUID site research coordinator will communicate (via email and or visits to ward and units) with nurse managers and inventory personnel when the bi-weekly inventory reports suggest that study fluid compliance is <80% for a specific area in the hospital; (3) the day before the study fluid swap from study period 1 to study period 2, an email communication will be sent to physicians, clinical directors, nurse managers and nurses to alert them to the swap and the date of initiation of study period 2; and (4) at the end of study period 2, a final email communication will be sent to physicians, trainees, nurse educators, managers and bedside nurses to let them know the trial is completed and to thank them for participating in FLUID.
Contamination and carry over
The risk of contamination due to inadequate adherence to the study fluid will be minimised through four mechanisms. (1) An automatic substitution order for the study fluid will be invoked during the trial study periods: nurses will be authorised by the senior management team at each participating hospital to perform an automatic substitution for the study fluid when the alternate fluid has been ordered by the treating physician. The authorisation mechanism for nursing to perform this automatic substitution will be determined by the senior administration at each hospital. The automatic substitution may be over-ridden if the treating physician indicates ‘no substitution’ in the physician’s orders. (2) The hospital ward shelves will be stocked with at least 80% study fluid for the duration of the study periods. (3) Bright signage prominently placed where 0.9% saline and Ringer’s lactate are stored will help to remind nurses about the automatic substitution. (4) The other resuscitation crystalloid fluid will be available only in small quantities (<20% will be available on the shelves of non-trial resuscitation crystalloid fluid).
The risk of carry-over (effect of study fluid on patients included in study period 1 carrying over to study period 2) is minimised as: the vast majority of patients will be different in each study period (average length of hospital stay in Ontario hospitals is 6.5 days)37; the majority of administered crystalloid resuscitation fluids occurs during the first few days of a hospital admission; and only index admissions will be included. Furthermore, a 1-week run-out (see figure 1) between study period 1 and at the end of study period 2 will be used to minimise the occurrence of patients being exposed to two different kinds of fluids during the same hospitalisation.
Intervention and control groups
The planned trial interventions are 0.9% saline (control fluid) and Ringer’s lactate (treatment fluid).
Both 0.9% saline and Ringer’s lactate with or without the addition of electrolytes will be stocked in the hospitals and administered in the usual way, that is, 500 or 1000 mL boluses or continuous intravenous infusions as specified by the treating physicians at the participating hospitals in an open-label fashion. The allocated study fluid will be the dominant fluid stocked (at least 80%) throughout the hospital for the duration of both study periods. Other fluids will not undergo substitution during the study periods (eg, 2/3 and 1/3, D5W, D5 half 0.9% saline and hypertonic saline).
Approach to safety
Both 0.9% saline and Ringer’s lactate are usual care resuscitation crystalloid fluids which have been in clinical use for decades and are used every day to resuscitate patients who are hypovolaemic, haemodynamically unstable and for the replacement of fluid losses. Despite their widespread use, no large multicentre randomised trials have been conducted to evaluate the effect of these two fluids on clinically important outcomes, and there remains diversity of crystalloid fluid use in clinical practice.38 Thus, participation in this trial poses no greater risk than that of routine care.
In advance of FLUID trial start-up at each participating hospital, several communication strategies will be implemented (see table 1 for more details) to ensure all key stakeholders (staff physicians, trainees and nurses) are educated about FLUID. With our communication strategies, we will also ensure that physicians and nurses know that there will be a small amount of the non-allocated study fluid available for use throughout the hospital if the treating physician chooses to opt out of using the study fluid for a given patient. Opting out may occur if the treating physician has a strong clinical reason to not use the allocated study fluid (eg, severe hyperkalaemia, severe metabolic alkalosis or acidosis, and severe brain injury).
An independent Data Safety and Monitoring Committee will conduct a blinded by group safety analysis of the FLUID primary clinical outcome (death or requirement for hospital re-admission at 90 days) after conclusion of the pilot trial and will advise the FLUID steering committee on continuing or stopping the clinical trial. Any serious adverse events considered related to the study fluid that are reviewed at morbidity and mortality rounds or reported to safety management committees at participating sites will be communicated to the site investigator who will then advise the FLUID study’s principal investigator.
Outcomes
Primary feasibility outcome
Adherence to the FLUID protocol
In the FLUID trial, adherence will be measured not at the individual patient-level, but according to the aggregate use of the study fluid throughout the hospitals (all hospital wards, monitored units and departments) using the hospital inventory system; monitoring adherence according to individual patients will not be feasible due to the sheer number of hospital admissions. To calculate adherence, the total use of the allocated study fluid will be divided by the total combined use of 0.9% saline and Ringer’s lactate.
Adherence will be monitored at 2-week intervals over the 14-week study periods. At the conclusion of the FLUID pilot trial, these adherence rates will be described according to each study group across all four participating hospitals, for individual hospitals, and for different wards across all and individual participating hospitals.
Logistical reasons (eg, stocking issues on the hospital wards, lack of study fluid availability in the hospital, lack of signage where fluids are stored indicating automatic substitution) for non-adherence will be documented at the participating hospitals to evaluate if non-adherence is modifiable from the logistical perspective.
Successful adherence to the FLUID protocol is defined as a total of at least 80% of the prescribed study fluid for each study group being administered across all four participating hospitals over the 3-month study periods.
Secondary feasibility outcomes
Time to research ethics board approval
Although FLUID meets ethical criteria for the use of a waiver of consent (see Ethics and Dissemination Section ahead), we recognise that different REBs may interpret justification for waiver of consent differently which may result in delays to REB approval. Delays in time to REB approval could hamper our ability to allocate sites at the designated time and complete the trial within the scheduled time period. Reasons for delays will be documented to understand whether the process could be modified to enhance the time to approval for participating hospitals in the large FLUID trial.
Successful time to REB approval will be defined as taking no >3 months (90 days) from REB submission to REB approval.
Time to readiness for study initiation
It is important to record and understand the time taken to initiate FLUID because minimising trial initiation delays may reduce the risk of sites dropping out and downstream complications related to trial operations. Delayed initiation of participating sites also tends to increase the operational costs of trials by increasing the overall duration of study conduct, which can have important implications when preparing the future trial budget. For the future large FLUID trial, randomisation will be blocked on time to reduce period effects. It would therefore be desirable for participating hospitals to be ready to initiate the trial in a similar time frame. Reasons for delays will be documented to understand if the process could be modified to enhance the time to readiness for study initiation in the large FLUID trial. Several operational issues that will be addressed before a site is ready for study start-up will be documented; these include education of nurses, physicians and trainees in advance of trial start up, ensuring availability of and stocking of study fluid throughout the hospital, placement of bright signage where fluids are stored throughout the hospital to remind nurses of the automatic substitution order.
Successful time to readiness for study initiation will be defined when a hospital takes no longer than 3 months (90 days) from REB approval to trial initiation.
Secondary clinical outcomes
All primary and secondary clinical outcomes for the future large FLUID trial will be described in aggregate (not by study group) in the pilot trial. The primary clinical outcome for the future large FLUID trial is a composite of death or re-admission to hospital within the first 90 days of the index hospitalisation; both outcomes are clinically important, relevant at the level of the healthcare system and to patients, and easily obtainable. Importantly, they have both been validated at ICES, are complete and highly accurate (≥99%).39 40 Secondary clinical outcomes include death and re-admission to hospital within the first 90 days of the index hospitalisation described as separate variables, requirement for dialysis, need for re-operation, need for re-intubation postoperatively, emergency department visits within the first 90 days of the index hospitalisation, length of stay in hospital and hospital discharge disposition.
Duration of treatment period, follow-up and data collection
A 14-week study period was selected because it was deemed a reasonable period for inventory control and turnover.
All follow-up and collection of data for enrolled patients at the participating hospitals will be captured through administrative health data that are housed at ICES. There will be no individual patient-level data collected by research coordinators in the participating hospitals. The clinical outcomes will be derived from provincial health administrative databases held at ICES. See the online supplementary file 2 for a summary of the data variables and the database sources from health administrative data housed at ICES.
Losses to follow-up
A strength of our study design is the ability to assess outcomes using administrative health data from the ICES with near 100% follow-up. We anticipate minimal losses to follow-up since all the clinical data and outcome measures from participating hospitals in Ontario are available at the ICES except for a very small number of patients who leave the province of Ontario within 90 days after enrolment. In a cluster crossover trial, hospital attrition could have substantial implications for power. However, since we will be able to analyse all randomised hospitals regardless of study completion under the intent-to-treat principle, we do not need to account for hospital attrition. In establishing the sample size of the large trial, we are planning to account for potential hospital withdrawal in the intent-to-treat analysis by adjusting the effect size in the sample size calculation, and we will implement strategies to minimise the risk of hospital withdrawal. The pilot study will include a debriefing after completing the pilot with participating hospital administrators, the heads of nursing and fluid inventory personnel to further understand challenges related to trial implementation that will inform the design of strategies to minimise hospital withdrawal in the larger future trial.
Sample size
Four hospitals will participate in the FLUID pilot trial, yielding data on at least 12 000 hospital admissions. Two critical parameters in sample size calculations for cluster crossover trials are the within-period intracluster correlation and between-period intracluster correlation coefficients (ICCs). The former is the correlation among patients in the same hospital and same period, while the latter is the correlation among patients from the same hospital but different periods. The between-period ICC is usually assumed to be less than the within-period ICC and their ratio is called the cluster autocorrelation coefficient (CAC) which dictates the strength of the correlation between cluster means over time.41 Sample size calculations also require specification of the primary outcome event rate in the control arm. A large number of sites would be required to estimate these coefficients and event rates with acceptable precision.42 We will be able to use population data from ICES across all eligible hospitals in the province of Ontario sites (rather than only four sites) to capture event rates and calculate correlation coefficients very accurately to inform the design of the large FLUID trial. For these reasons, the sample size for this pilot is not based on precision or power considerations, but instead, on logistical and feasibility considerations within the constraints of a pilot study.
We used the methodology of Hooper et al 43 to conduct preliminary sample size calculations for the large trial for a range of plausible sample size parameters. Our preliminary calculations show that between 6 and 24 sites in a two-period three-repeated measures per period cluster crossover trial would be required to achieve 80% power to detect a clinically important absolute difference of 1% in the composite outcome of death or requirement for re-admission to hospital within the first 90 days of the index hospital admission, assuming a control arm (0.9% saline) proportion of 0.16, a range of within-period ICCs between 0.001 and 0.01, and a range of cluster autocorrelation coefficients from 0.75 to 1, with an average of either 1000 or 1500 patients per hospital per month. These calculations will be refined following the successful completion of the pilot and will need to be inflated to account for non-adherence, an approximate small sample correction, and the use of the normal approximation to the binomial distribution, cluster size variation and any potential cluster attrition. The results of the preliminary sample size calculations are summarised in table 2.
Preliminary sample size calculation for the large FLUID trial for a range of plausible sample size parameters
Analytical plan
In this pilot trial, analyses will primarily involve descriptive statistics. All feasibility outcomes (eg, protocol adherence and time to REB approval) will be described at each site and across all sites. The event rates for the primary and secondary feasibility and clinical outcomes will be described using frequencies and proportions together with 95% two-sided CIs, in addition to event rates calculated across the population of hospitals using Ontario provincial administrative health data housed at ICES. Within-period and between-period ICCs for the primary composite clinical outcome measure for the large FLUID trial will be estimated across the population of hospitals to inform the sample size calculation for the large trial. The trial and intervention costs will be also estimated.
Subgroup analyses
Several predefined subgroups will describe our clinical outcomes among patients who are more likely to receive higher exposure to fluids, with greater risk profiles or higher severity of illness. These include age (<18, 18 to ≤65, 66 to ≤80, and >80); sex; type of hospital admission (medical and surgical), trauma admissions, sepsis admissions; elective versus urgent/emergent surgery; surgical subgroups (general, thoracic, cardiac, vascular, orthopaedic, spine, obstetrics, gynaecological, ear, nose, throat, plastics, urological and other); and admission to an ICU. In the FLUID pilot trial, the clinical outcomes according to these subgroups will be described in aggregate (not according to study groups).
Sensitivity analyses will describe the clinical outcomes in patients who are extremely unlikely to receive the study fluids (direct hospital admissions to psychiatry) and those who are otherwise healthy (patients who have vaginal and caesarean births and patients who are admitted electively for day surgery defined by a hospital admission of <24 hours).
Trial management
The lead site investigators, the principal (LM), co-principal investigators (MT and DF) and the FLUID study manager (TM) will be responsible for fielding study questions at their hospitals during the conduct of the trial. Inventory personnel will ensure all shelves are adequately stocked for the study period duration and ensure bright signage to indicate use of the study fluid throughout the hospital. Inventory personnel will send electronic reports to the coordinating centre bi-weekly to monitor compliance to the study intervention for the trial duration. If compliance is <80%, the coordinating centre will inform the site to see if low compliance is potentially modifiable (study fluid not stocked adequately in the different geographic regions, requirement for additional education about FLUID and so on). At the conclusion of the trial, the principal investigator (LM) and study manager (TM) will conduct structured debriefing sessions with key stakeholder groups to discuss challenges during pilot trial implementation to help ensure the successful conduct of the future large FLUID trial.
Patient/public involvement
In the early planning phases of the FLUID pilot trial, we recruited a patient partner (AM) who is an active member of our FLUID executive committee. Our patient partner participates on FLUID executive conference calls and contributes to all decisions about the study. Our patient partner participated in and contributed to discussions about the FLUID pilot trial study design, outcome measures and ethical argumentation, and the communication strategies related to the trial. In the early planning phases of the pilot trial, our team also consulted with The Ottawa Hospital’s Patient Advocacy Committee to guide our communication approach to hospitalised patients and their family members about this waived consent study.
Ethics
We have had extensive discussions about ethical issues within our FLUID team in preparation for the FLUID pilot trial. According to recommendation 1 of the ‘Ottawa Statement on the ethical design and conduct of cluster randomised trials’,44 the choice of cluster (as opposed to individual) randomisation should be carefully justified. Our justification is provided in the Study Design Section. In brief, we have selected a cluster randomised trial as the intervention can only feasibly be evaluated when implemented at the hospital level (ie, a cluster-level intervention); it is essential to minimise contamination and maximise compliance by having the same study fluid available throughout the hospital. Randomisation of individual patients would be logistically challenging and very costly.
We will seek research ethics approval from all participating sites as per recommendation 2 of the Ottawa Statement. Cluster-level interventions present challenges in seeking individual patient informed consent. For these reasons, we will ask the REBs to approve waivers of patient consent for both study interventions and collection of clinical data that is housed at ICES. As per recommendations 6 and 7 of the Ottawa Statement, as well as the Tri Council Policy Statement for the Ethical Conduct for Research Involving Humans,45 a waiver of consent may be justified when the risks of study participation are minimal and requiring informed consent would render the study infeasible. See the online supplementary file 3 for a detailed discussion on the rationale and justification for a waiver of consent.
Study timeline and dissemination
The FLUID pilot trial was approved by the Ottawa Health Science Network Research Ethics Board 9 June 2016 (OHSN-REB approval #: 2015 0619-01 hours). The first FLUID pilot site began recruitment on 15 August 2016 and it is anticipated that it will be completed in approximately 2 years to account for the sequential study design. We estimate 6 months to obtain REB approval and for study preparation for each of the participating hospitals, 10 months to complete both study periods at all four sites, 2 months for site debriefing sessions with key stakeholders from participating sites after trial completion, 3 months for ICES to prepare and conduct the primary and secondary outcome event estimates, and 3 months for manuscript preparation and submission for publication.
Discussion
The FLUID trial design is innovative in its use of a cluster crossover design, waivers of patient informed consent to include all hospitalised patients and the use of routinely collected health data to determine study outcomes. Our pilot trial will establish the feasibility of conducting the large trial using this design and provide essential information to optimise the design, implementation, conduct and successful completion of the large trial.46–48 The pilot trial will inform the development of strategies to overcome barriers related to hospital recruitment, research ethics board approval processes, trial start up and operations, protocol adherence and retention. Finally, by extending the ICES data creation plan from the pilot sites to the larger population of eligible sites within Ontario (at no extra cost), we have a unique opportunity to obtain reliable estimates of the event rates as well as the within-period and between-period ICCs—two essential parameters required in power calculations for cluster crossover trials.
If our feasibility objectives are not met, then we will need to re-evaluate the protocol and plans for the larger trial in collaboration with our FLUID executive team and the Canadian Critical Care Trials Group and only consider moving toward the large trial if we have sufficiently addressed our challenges. If we deem the pilot trial to be feasible, then data will be included in the large trial (ie, this will be an internal pilot). If substantial modifications to the protocol are required before progressing to the large trial, then data from this pilot will not be included in the large trial (ie, this will be considered an external pilot). Regardless of the next steps, lessons learnt from this pragmatic pilot trial are likely to inform other hospital-wide cluster crossover randomised trials in the future.
Acknowledgments
The authors would like to thank the Canadian Critical Care Trials Group (CCCTG) for their assistance with the design of the FLUID pilot trial. They would also like to thank Jan Freidrich from the CCCTG for his critical review of the FLUID protocol manuscript.
References
Footnotes
Contributors LM, MT and DAF: conceived the project idea. LM: created the initial draft of the manuscript. All authors contributed to the development of the trial protocol; critical revisions and approved the final version of the manuscript.
Funding The FLUID pilot trial was funded by the Canadian Institutes for Health Research and the TOHAMO Innovation Fund Competition who did not contribute to the design of the FLUID pilot protocol.
Competing interests None declared.
Ethics approval Ottawa Health Science Network Research Ethics Board.
Provenance and peer review Not commissioned; externally peer reviewed.