Article Text

Abstract
Introduction Dopamine dysregulation has been identified as a key modulator of behavioural impairment in neurofibromatosis type 1 (NF1) and a potential therapeutic target. Preclinical research demonstrates reduced dopamine in the brains of genetically engineered NF1 mouse strains is associated with reduced spatial-learning and attentional dysfunction. Methylphenidate, a stimulant medication that increases dopaminergic and noradrenergic neurotransmission, rescued the behavioural and dopamine abnormalities. Although preliminary clinical trials have demonstrated that methylphenidate is effective in treating attention deficit hyperactivity disorder (ADHD) symptoms in children with NF1, its therapeutic effect on cognitive performance is unclear. The primary aim of this clinical trial is to assess the efficacy of methylphenidate for reducing attention deficits, spatial working memory impairments and ADHD symptoms in children with NF1.
Methods and analysis A randomised, double-blind, placebo-controlled trial of methylphenidate with a two period crossover design. Thirty-six participants with NF1 aged 7–16 years will be randomised to one of two treatment sequences: 6 weeks of methylphenidate followed by 6 weeks of placebo or; 6 weeks of placebo followed by 6 weeks of methylphenidate. Neurocognitive and behavioural outcomes as well as neuroimaging measures will be completed at baseline and repeated at the end of each treatment condition (week 6, week 12). Primary outcome measures are omission errors on the Conners Continuous Performance Test-II (attention), between-search errors on the Spatial Working Memory task from the Cambridge Neuropsychological Test Automated Battery (spatial working memory) and the Inattentive and Hyperactivity/Impulsivity Symptom Scales on the Conners 3-Parent. Secondary outcomes will examine the effect of methylphenidate on executive functions, attention, visuospatial skills, behaviour, fine-motor skills, language, social skills and quality of life.
Ethics and dissemination This trial has hospital ethics approval and the results will be disseminated through peer-reviewed publications and international conferences.
Trial registration number ACTRN12611000765921.
- clinical trials
- neurogenetics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first randomised controlled trial evaluating the effects of methylphenidate in the treatment of cognitive deficits in neurofibromatosis type 1 (NF1).
This study is adequately powered to provide a clinically meaningful outcome.
The cognitive primary outcome measures are analogous to methylphenidate-responsive tests previously used in NF1 mouse models, assessing sustained attention and spatial working memory.
A multimodal approach including event-related and resting state functional neuroimaging will investigate the effects of methylphenidate on neurobiological processes in NF1 and how these relate to cognitive and behavioural changes.
A 6-week intervention period may not be sufficient to see the full impact of treatment on daily functioning and quality of life; open-label extension studies will be required to determine these long-term effects.
Introduction
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder that is caused by a diverse range of loss-of-function mutations in the NF1 gene, which resides on chromosome 17q11.2. With a birth incidence of 1 in 2700, NF1 is one of the most common monogenic disorders affecting cognitive function.1 The NF1 gene encodes the protein neurofibromin, a tumour suppressor that is a negative regulator of Ras2 and a positive regulator of dopamine (DA) homeostasis.3 Loss of neurofibromin expression results in increased Ras activity and cell growth.4 Diagnostic features include alterations in skin pigmentation (café au lait spots, skinfold freckling), Lisch nodules and nervous system tumours (neurofibromas and optic pathway gliomas).5 While typically regarded as a cancer predisposition syndrome, the most common complication of NF1 in childhood is impairment in academic achievement and cognition, with attentional, executive, language and visuospatial functions frequently affected.6–9 Attentional problems are one of the most commonly reported impairments, with up to 70% of children displaying deficits in one or more aspects of the attention system (sustained, selective, divided and shifting attention).6 10 11 Significant executive impairments are also reported, including problems with working memory and inhibitory control.12–14 The NF1 cognitive phenotype has many similarities to that seen in children with attention deficit hyperactivity disorder (ADHD) and over one-third of children with NF1 meet diagnostic criteria for ADHD.14–16 Like children with idiopathic ADHD, where dissociations between cognition and symptoms have been established,17 18 disruption to attentional and executive functions are not tightly bound to clinical symptoms, with commensurate cognitive deficits occurring in children with NF1 irrespective of ADHD comorbidity.12 16 Functional MRI (fMRI) studies in children with NF1 reveal disturbances within neural networks associated with working memory,19 inhibitory control20 and attention,21 which further resemble findings in children with idiopathic ADHD.22–26 In particular, hypoactivation in prefrontal regions,19 20 striatum19 and anterior cingulate cortex21 have been identified as loci of neurobiological dysfunction in NF1. Critically, executive and attentional deficits, as well as ADHD symptoms, significantly impact the scholastic abilities16 and social competence27 28 of children with NF1, highlighting them as key clinical targets for therapeutic intervention.
Various genetically engineered Nf1 mouse strains have successfully modelled the cognitive and behavioural deficits seen in children with the disorder, enabling investigation into the underlying pathophysiology and identification of new therapeutic drug targets. Mice with a heterozygous inactivating mutation in the NF1 gene (Nf1 +/-) demonstrate Ras signalling-dependent increases in gamma-aminobutyric acid in the brain.29 Spatial learning and attention impairments have been associated with elevated Ras signalling and inhibition of Ras transforming activity with lovastatin has rescued the behavioural phenotype in Nf1 mice.30 Despite the significant promise, human randomised controlled trials (RCTs) using statins have demonstrated poor efficacy, with limited treatment effects on cognitive outcomes.31–33
Convergent mouse and human data also indicate that reduced neurofibromin expression can lead to dysregulated neuronal DA levels, which may contribute to the observed attentional and learning impairments in children with NF1.34–37 Nf1 +/- mice with bi-allelic inactivation of Nf1 in GFAP+ astroglial cells display reduced exploratory behaviours, attention abnormalities36 and spatial learning deficits.35 These behavioural deficits have been linked to a presynaptic DA defect in the striatum36 and hippocampus.35 In vivo experiments using positron emission tomography imaging with 11C-raclopride have extended these research findings demonstrating increased striatal binding, consistent with abnormally low DA levels.34 Treatment with methylphenidate (MPH), a stimulant medication that increases extracellular DA availability by inhibiting its reuptake by the DA transporter, corrected mice behavioural abnormalities and restored the DA levels in the striatum and hippocampus.34–36 These findings, which link NF1 gene expression, DA dysregulation and attention deficits provide a robust foundation for the evaluation of DA reuptake inhibitors such as MPH as a potential treatment for both cognitive and behavioural deficits in children with NF1.
In idiopathic ADHD studies, MPH is effective at reducing ADHD symptoms with a large effect size (0.8–1.0).38 39 fMRI studies in ADHD participants indicate that acute doses of MPH upregulate and normalise under functioning brain regions in ADHD, including the basal ganglia and prefrontal cortex.40 41 While the effects of MPH on cognition in ADHD are less clearly understood, a meta-analysis of 36 studies reported significantly improved executive memory, non-executive memory, reaction time, reaction time variability and response inhibition in MPH versus placebo conditions, although with smaller effect sizes than those reported for ADHD symptoms.42
Clinical trials of MPH in NF1 are limited. An open-label uncontrolled trial of MPH conducted in 20 children with NF1 who met diagnostic criteria for ADHD suggested improved performance on a computerised task of attention and corresponding gains on informant-reported behaviour (attention, anxiety/depression and social competence) across a 12-month treatment period.14 The only other clinical trial evaluating the effects of MPH in children with NF1 reported beneficial effects of MPH in reducing parent-reported ADHD symptoms in children with NF1 following 4 weeks of treatment.43 Despite the preclinical evidence that DA-targeted therapies may improve cognitive deficits in children with NF1, there are no published RCTs evaluating the effect of MPH on cognitive outcomes in this population.
Methods and analysis
Study aims
The primary aim of this study is to determine the efficacy of MPH at improving sustained attention, spatial working memory and ADHD symptoms in children with NF1. The secondary aim is to evaluate the effects of MPH on visuospatial learning, executive functioning, other aspects of attention, expressive language, social skills, fine-motor skills, quality of life and behaviour.
Exploratory aims of the study are to examine the associations between MPH-related changes in the various aspects of cognition and changes in ADHD symptoms. We will also explore the effect of MPH on functional brain activity in children with NF1 and relate this to improvements in cognitive and behavioural function.
Study design and setting
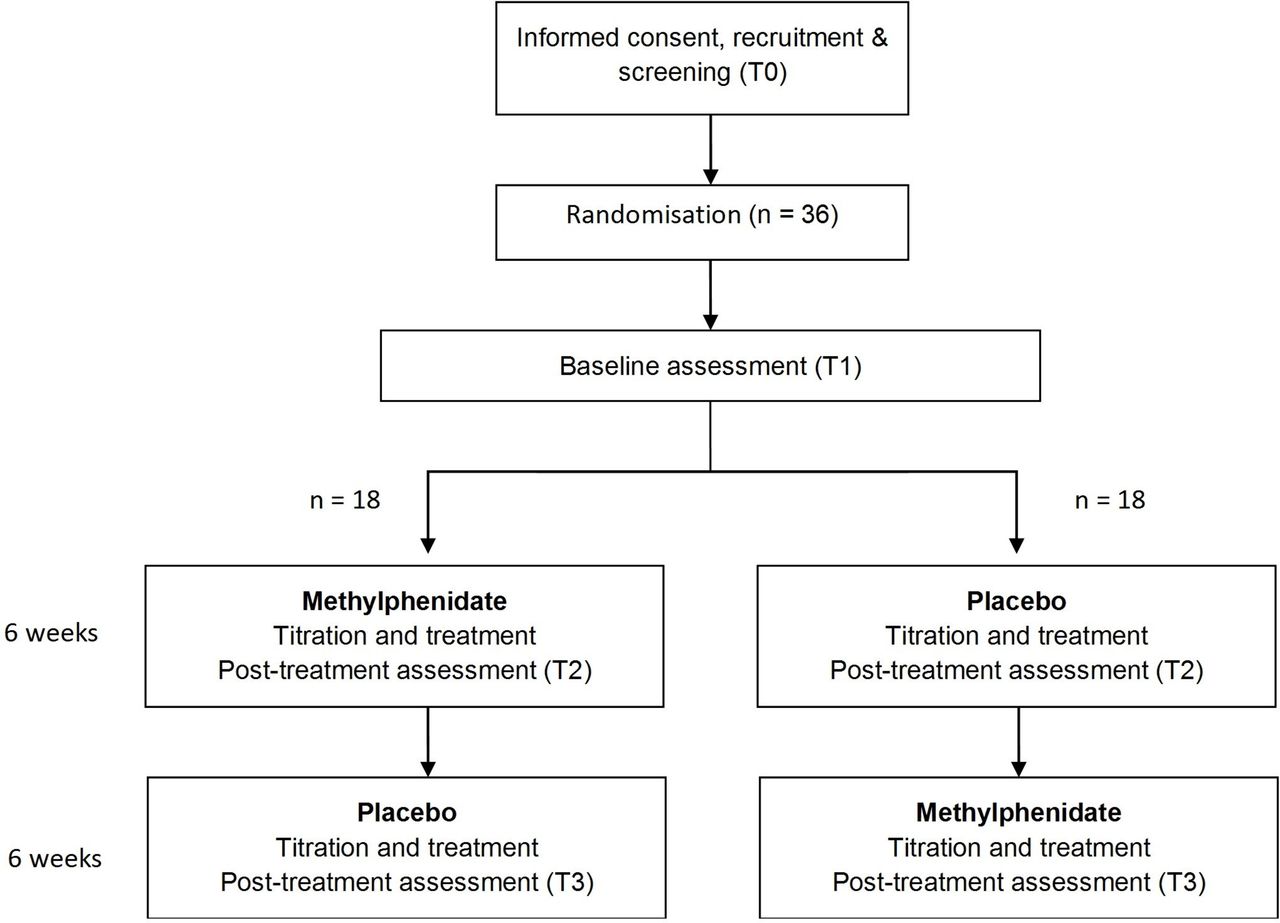
A phase II, multicentre, randomised, double-blind, placebo-controlled clinical trial with a two period crossover design. Participants will be randomised to sequence A (6 weeks of MPH followed by 6 weeks of placebo) or sequence B (6 weeks of placebo followed by 6 weeks of MPH). Cognitive testing, behavioural outcomes and neuroimaging be completed by participants at baseline and at the end of each treatment condition. Figure 1 provides an overview of the study.

Flow diagram of participant allocation.
Study procedures
Prospective participants will be recruited from outpatient NF1 clinics at The Children’s Hospital at Westmead (CHW) in Sydney and The Royal Children’s Hospital in Melbourne, Australia. Children are referred to these clinics by general practitioners and medical specialists for evaluation, diagnosis and management of NF1. Participant recruitment will take place via the following methods: (1) families of patients attending NF1 clinics will be asked whether they would be willing to hear more about the trial, (2) mailing information packages to families that attend the clinics and (3) online announcements on the Australian Children’s Tumour Foundation website. The research team will contact families interested in participating and provide further study information and address any questions about the trial. Those willing to participate will be asked to provide written informed consent.
Screening and eligibility
Eligibility will be checked at a preliminary screening appointment (see ’Inclusion criteria' and ’Exclusion criteria' sections below). At this visit, the site physician will collect a detailed medical history including an intake interview with questions aimed to identify eligibility of the participant. The physician will also conduct a physical examination to confirm NF1 diagnosis and document height, weight, blood pressure, pulse rate and head circumference. Baseline adverse effects will be documented, allowing us to determine whether subsequently endorsed adverse effects are related to study medication. All participants will complete a screen for cardiac abnormalities (ECG), and eligible females will complete a urine pregnancy test. Concomitant medications taken by the participant at commencement of the trial will be documented. Parents/caregivers of all participants will undergo a structured interview using the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-Kid) to screen for psychological disorders and substance abuse.44 Parents/caregivers will also complete the Conners 3-Parent45 to identify the nature and severity of ADHD symptoms at baseline. The study psychologist will determine whether the participant satisfies cognitive-based entry criteria by administering the Conners Continuous Performance Test II (CPT II) (sustained attention),46 Cambridge Neuropsychological Test Automated Battery Spatial Working Memory (CANTAB SWM)47 and the Wechsler Abbreviated Scale of Intelligence (WASI; IQ).48 If the participant has completed an IQ test within 12 months of screening, then the WASI will not be administered and the previous score will be used. Examples of acceptable IQ tests include the WASI, Wechsler Intelligence Scale for Children—Fourth Edition49 or Fifth Edition50 and the Stanford-Binet Intelligence Scales Fifth Edition.51 Participants will be informed of the potential risks and benefits of participating, the aim of the study and that participation is voluntary. The first participant was randomised to study on 30 July 2011 and we anticipate the end date for enrolment to be December 2019.
Inclusion criteria
Diagnosis of NF1 based on NIH criteria.52
Males and females aged between 7 and 16 years of age (inclusive) at time of enrolment.
A full scale IQ≥70 or, if a significant discrepancy is present between perceptual reasoning index and verbal comprehension, the higher index must be ≥70.
An abnormal Inattentive or Hyperactivity/Impulsivity Symptom Scale on the parent-rated Conners 3, defined as a T score ≥65.
Baseline performance of one or more SD below the population mean on the CPT-II omissions subscale and/or the CANTAB SWM task total between-search error score.
Exclusion criteria
Insufficient English to understand the cognitive assessments or comply with the study protocol.
Intracranial symptoms or pathology such as epilepsy, hydrocephalus, diagnosed traumatic brain injury or progressive intracranial tumours that may impact cognitive and behavioural function (children with asymptomatic or static lesions will be eligible).
Significant visual and/or hearing problems that, in the view of the site principal investigator, significantly impact on the validity or reliability of cognitive testing.
Blood pressure measurements (systolic or diastolic) ≥95th percentile for age, sex and height at screening or a diagnosis of hypertension.
An abnormal ECG result at the time of screening deemed clinically significant by study physician.
Known contraindication or intolerance to the use of stimulant medications.
Use of any investigational drugs within 30 days of screening.
The current use of prescription or over-the-counter medication to induce sleep.
Unable to swallow capsules.
Pregnancy.
Known past or present substance abuse or dependence, including alcohol, as determined by the MINI-Kid.
Presence of any other comorbid psychiatric or psychological disorder (excluding ADHD, oppositional defiant disorder, conduct disorder and pervasive development disorder/autism spectrum disorder) including depressive disorder, anxiety disorder, psychotic disorder, suicidality, Tic disorder, anorexia or bulimia nervosa.
Treatment with MPH or any other stimulant medication in the past 21 days.
Randomisation and blinding
Eligible participants will be randomised into either sequence A (MPH followed by placebo) or sequence B (placebo followed by MPH). Randomisation will be managed by an independent statistician at CHW. Randomisation will be performed using computer-generated random number allocation tables based on a 1:1 allocation between groups. Randomisation codes will be maintained in a password-protected file and shared with the clinical trial pharmacy at each site. The clinical trial pharmacy will present the MPH and placebo capsules in plain bottles that do not contain treatment allocation. The clinical trial coordinator will collect the treatment and distribute to the participant. Participants, study physicians, clinical trial coordinators, researchers and investigators will be blind to treatment allocation. The password-protected randomisation allocation file will only be accessible in an event where unblinding is deemed necessary, such as developing a concurrent medical condition that might preclude or contraindicate the administration of MPH.
Intervention
The active treatment is MPH, a central nervous system stimulant. MPH is a racemate consisting of a 1:1 mixture of d-threo methylphenidate and l-threo methylphenidate. Although its mode of action in humans is not completely understood, MPH presumably exerts its stimulant effect by an inhibition of DA and norepinephrine reuptake, primarily in the striatum, and in other regions, such as the prefrontal cortex.53 54 New South Wales health guidelines indicate the dose of MPH to treat children with ADHD should not exceed 60 mg/day and should be no more than 2 mg/kg/day. Given the cohort in the current study is a new population, we have adopted a maximum dose of up to 1.5 mg/kg/day, up to 50 mg/day. Capsules are compounded by Stenlake Compounding Pharmacy or CHW clinical trial pharmacy using identical methods and will contain either 5 mg MPH, 10 mg MPH or inactive lactose powder (placebo). MPH and placebo capsules will be indistinguishable and provided to participants as crushed powder encased in opaque gelatin capsules.
Titration and dose optimisation
A prescription requesting the number of capsules for the titration phase (based on the weight of the participant) will be provided to the site pharmacy on enrolment. The study coordinator will collect the drug from the site’s clinical trial pharmacy and distribute it to participants.
Treatment dose will be individually determined during a titration phase, which will be identical for both placebo and MPH conditions. Participants will be provided with a titration schedule according to their baseline weight. The schedule will include detailed instructions outlining the dosage and timing for the titration phase. A member of the research team will discuss these instructions with the participant’s family to ensure comprehension. Participants will commence with one capsule administered in the morning on day 1 and will slowly escalate to a maximum of three administrations per day to the maximum dose allowed for a child’s weight (1.5 mg/kg, ≤50 mg/day). Maximum doses are rounded down to the nearest 5 mg. Participants will be instructed to swallow the capsules whole following food and to take them at the same time every day with approximately 4 hours between doses.
The optimal treatment dose will be determined individually based on the presence of adverse effects using the Side Effects Rating Scale (SERS),55 which is a parent-rated scale that assesses the frequency and severity of 17 common adverse effects associated with stimulant medication from 0 (absent) to 9 (severe). To do this, participants and their families will be asked to keep a daily record of any adverse effects using a medication diary. During the titration phase, study personnel will contact the family via telephone at the mid-point (week 1) and end (week 2) to assess adverse effects using the SERS. If a participant scores 7 or above on any of the 17 SERS criteria, the child’s dose will be reviewed by the site physician. The physician will ascertain which day the adverse effect first occurred based on the medication diary. The participant’s dose will remain at the level at which the adverse effect was first experienced. The family will complete the SERS again during a follow-up phone call two days later. If the adverse effect remains ≥7, then the treatment dose will be reduced to the previous dose at which the significant adverse effect was not observed. If significant adverse effects are not reported at the reduced dose then this dose level will be considered the participant’s optimal daily dose for the remainder of the treatment period. If the significant adverse effects persist on the reduced dose, the dose reduction cycle will be repeated until the SERS symptom is rated <7.
The SERS will be administered at several time points during each treatment condition. The site physician will make a clinical judgement on whether there has been a significant increase to the existing problem based on parent observations and the SERS baseline measurement. If deemed a significantly increased adverse effect, the dose reduction procedure will apply.
To assess compliance, participants will be required to record the time and number of capsules taken in a medication diary. Unused capsules will be returned at the end of each treatment period and the number of returned capsules will be recorded.
Once the first treatment condition has been completed, participants will cross over to the other treatment on the following day. There will be no washout period. The procedures for the second treatment condition will be identical to those for the first.
Cognitive and behavioural outcomes
The schedule of outcome assessments is detailed in figure 2. Primary and secondary outcome measures will be administered at baseline (T0/T1) and following completion of each 6-week treatment condition (±3 days T2, T3). Since MPH is rapidly absorbed56 and the pharmacodynamics effects mirror the pharmacokinetic profile, cognitive outcomes will be administered 45 min after treatment dose.57 Parents and teachers will be instructed to rate symptom questionnaires based on behaviour over the previous 4 weeks.

{kind=link}
{kind=link}
Schedule of enrolment, interventions and assessments Standard Protocol Items: Recommendations for Interventional Trials figure.
Primary outcome measures
All primary outcomes capture high basal rates of impairment in NF1.11 12 33 58 59 Cognitive measures are analogous to MPH-responsive tests previously used in Nf1 mice,34–36 assessing sustained attention and spatial working memory. The symptom rating scale is standard for ADHD research.
Sustained attention will be measured using the CPT-II: a well-established computerised measure with a significant history of use in identifying attentional deficits and the effects of stimulant medication in research involving individuals with idiopathic ADHD.60 61 Omission errors will be the primary outcome variable from this test.
Spatial working memory will be measured using the CANTAB SWM task.47 The SWM task assesses a subject’s ability to retain spatial information and to manipulate remembered items in working memory. The primary outcome variable from this assay will be total between-search errors.
ADHD symptoms will be measured by the Conners 3-Parent questionnaire,45 a published and validated tool for assessing the presence of inattentive and hyperactive/impulsive behaviours in children and adolescents. Both the Inattentive and Hyperactive/Impulsivity Symptom Scales will be used as primary outcomes. Parents will be instructed to base their ratings on the participant’s behaviour over the previous 4 weeks.
Secondary outcome measures
To understand the potential benefits of MPH on a wider range of cognitive and behavioural outcomes, a broader battery of tests are included as secondary end points. Secondary outcome measures include the Test of Everyday Attention for Children (TEA-Ch)62 to measure selective attention (Sky Search), sustained attention (Score!), divided attention (Sky Search Dual Task) and attentional control/switching (Creature Counting): the CANTAB Stockings of Cambridge Task and Paired Associate Learning Task47 to respectively measure spatial problem solving and spatial learning: the Grooved Pegboard63 to measure fine motor skills: the Judgement of Line Orientation (JLO) task64 to measure visuospatial perception and orientation and Formulated Sentences from the Clinical Evaluation of Language Fundamentals—Fourth Edition Australian Version65 to measure expressive language. Commission errors from the Conners CPT-II will also be a secondary end point of response inhibition.
In addition to the neurocognitive outcomes, a number of parent-rated questionnaires will also be administered including the Behaviour Rating Inventory of Executive Function66 as a measure of everyday executive behaviour: the Social Skills Improvement System Rating Scales67 to evaluate social skills: the Child Behaviour Checklist (6–18)68 to assess children’s internalising and externalising behaviours and the Paediatric Quality of Life Scale 4.0 Generic Core Scales69 to measure health-related quality of life (both parent and self-report). Where possible, the Conners 3 Teacher will also be collected to measure ADHD symptoms at school. Secondary outcome measures are published assessment tools that are widely used in research settings and have standardised instructions minimising between-site variation. Alternate test versions will be administered at the second assessment where available (TEA-Ch and JLO).
Neuroimaging outcomes
Neuroimaging will be performed at baseline and at the end of both treatment periods (week 6 and week 12). Imaging will occur within the therapeutic window of MPH; approximately 45 min after administration. The time of day the participant is scanned (following morning, mid-day or afternoon dose) will be consistent across assessment visits. Neural correlates of cognitive fMRI tasks and the default mode network will be assessed after placebo and MPH treatment in children with NF1.
Using event-related MRI procedures, participants will perform tasks in the MRI scanner that assess various cognitive functions. This will enable us to determine the degree to which MPH is able to influence abnormal functional neural networks. We will use a well-established Auditory Oddball Task to probe the neural correlates of selective attention; a Continuous Performance Task to investigate neural mechanisms of sustained attention and working memory and a classic Go-NoGo Task to examine the neural process of inhibitory control. The total scan time for each paradigm is standardised to 5 min and 8 s. Details of the fMRI paradigms have been published,70 and we have established abnormal NF1-related activation in fronto-parietal and fronto-striatal networks using the Oddball and Go-NoGo paradigms.20 21
Participants will also undergo a resting state (rs)- fMRI sequence lasting approximately 5 min. Participants will be instructed to lie in the scanner with their eyes closed for the duration of the scan. This sequence will be used to identity MPH versus placebo-induced changes in functional interactions between neural networks at rest. A three-dimensional T1-weighted MRI will be obtained from all participants.
Adverse event monitoring
Routine monitoring will occur at screening (T0), week 6 (T2) and week 12 (T3). Blood pressure, pulse measurements and growth parameters (weight, height) will be recorded during monitoring visits. Adverse events (AEs) will be collected and recorded at monitoring visits and will be based on signs and symptoms reported by the participant/parent/caregiver or observed by the investigator during the physical examination. SERS symptoms will also be recorded as AEs. Documentation for all AEs will include the specific event/condition, the dates and times of occurrence, the event severity, duration, likely relationship to MPH, action taken and date of resolution. Severity of AEs will be graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (V.3; see table 1). Grade 1 and grade 2 AEs will be considered non-serious AEs. Grades 3–5 are considered serious AEs. If medication is used to treat the AE, the dose, route of administration, date and time medication is provided, and an indication whether the medication will be continued will be required. If a participant experiences a grade 2 AE or serious AE which has a possible relationship to study drug during any stage of the trial, their current dose will be reviewed by the site physician and may be withheld or modified at the physician’s discretion. Any change in dose will be documented.
Categorisation of adverse effects
All serious AEs will be reported to the Human Research Ethics Committee that approved the study within 72 hours. An independent Data Safety Monitoring Board will evaluate all AEs at regular intervals. A project management group will meet regularly to monitor the progress of the trial. This group will include the Chief Investigator, site principal co-investigators and researchers.
Sample size
Our study is powered for the primary efficacy analyses. Primary outcome measures are SWM between search errors (CANTAB), CPT-II omission errors and the Inattention and Hyperactivity/Impulsive Symptom Scales from the Conners 3. A previous open-label study, which examined the effects of stimulant medication on attention in children with NF1 using a different continuous performance task, reported an effect size of 1.24 for omission errors following MPH treatment for children with NF1.14 Similarly, a previous RCT in children with NF1 examined the effect of MPH on Conners Parent ADHD Rating Scale scores and reported an effect size of Cohen’s d=0.96.43 Based on these previous studies in children with NF1, we have estimated an effect size of d=0.96, which would be classified as a treatment effect of large to very large magnitude.71 To detect an effect size of d=0.96 in a mixed model crossover design with 80% power and a two-tailed significance of 0.05, a total sample size of 36 is required.72
Statistical analysis
Continuous outcome measures will be analysed using an intention-to-treat, mixed-effects linear model. Placebo and MPH data points will be compared, adjusted for baseline data and any potential carryover effects and clustered at both the individual site level. The mixed model allows for more complex within-subject correlation structures, and accommodates missing and unbalanced data between randomisation sequences. Therefore, within this model, it is not necessary to impute values for missing data. Effect sizes will be calculated as the mean difference between MPH and placebo divided by the SD of the differences (presented as Cohen’s d). All secondary outcome measures will be analysed using the same technique.
Patient and public involvement
Neither patient nor the public were involved in the development of the research questions, selection of outcome measures, study design or study conduct.
Ethics and dissemination
Ethical considerations
This clinical trial has been approved by the Human Research Ethics Committee of the Sydney Children’s Hospitals Network (HREC16/SCHN/261). Governance approval of local ethics has been obtained at both study sites. Any protocol modifications will be communicated to the study team, ethics committees and the trial registry. The study will be conducted in compliance with the guidelines for Good Clinical Practice and the Declaration of Helsinki and the National Health and Medical Research Committee National Statement on Ethical Conduct in Human Research. Written informed consent will be obtained from all participants before participation. During the informed consent process, a member of the research team will provide information about trial including but not limited to the study objectives, potential risks and benefits, inconveniences and the participants’ rights and responsibilities. Any questions about the trial will be addressed in detail. As participants are minors, written informed consent will be obtained from their parent/legal guardian. In addition, a child-friendly written information sheet will be provided and verbal agreement for participation will be obtained from the child. Participation will only proceed if the child or adolescent agrees to participate in the research. This trial is registered with the Australian and New Zealand Clinical Trial Register (ACTRN12611000765921).
Data management and confidentiality
Data management will be coordinated from The Children’s Hospital at Westmead and entered into the study database. Quality control will be implemented, including checks for double data entry and using range checks data values. All data will be anonymised and stored by secure means. All paper charts, forms and information associated with the study will be kept in a locked cabinet in lockable offices at each site’s buildings which have swipe access and security presence. All participants will be assigned a unique ID that will be used throughout the study. All data stored in computer systems will be password protected and stored using the participants' ID. A list of participant names and IDs will be kept separately on the password-protected principal investigator’s computer. Members of the research team involved in the analysis will have full access to the de-identified final study dataset.
Dissemination
The results of this trial will be submitted for publication in peer-reviewed journals and the key findings presented at national and international conferences. Members of the study team will also disseminate study results at patient seminars and parent groups. The investigator team will write all articles submitted for peer-reviewed publications and authorship inclusion and order will be guided by levels of contribution.
Discussion
Preclinical trials suggest that DA-targeted therapies, such as MPH, may be useful treatments for children with NF1-associated cognitive abnormalities. To date, previous studies have reported a beneficial effect of MPH on ADHD symptoms in children with NF1,14 43 and the one trial that examined the effects of MPH on child-direct assessments, suggested a robust treatment response for attention over a 12-month period.14 To our knowledge, this is the first RCT evaluating the effects of MPH on cognition in NF1 and will provide important evidence for the efficacy of MPH in treating deficits in attention, working memory and ADHD symptoms in children with NF1 over a 6-week period. Open-label extension studies will be required to determine long-term effects of more distal outcomes such as social functioning, academic achievement and psychiatric comorbidities.
References
Footnotes
Contributors NAP, KNN, JMP and BB developed the original concept of the trial and drafted the original trial protocol and methodology. SJCH, PH, MSK and DRC provided additional advice on the trial design and statistical methods. DRC, SM and MR commented on the final preparation of the protocol. All authors read and approved the final manuscript.
Funding This work is supported by the Children’s Tumor Foundation (grant number 2012-10-003).
Competing interests DRC has received grants and personal fees from Shire Pharmaceutical Company and Vifor Pharma; personal fees from Janssen-Cilag, Eli Lilly, Novartis, Flynn Pharma, Medice Arzneimittel Pütter and Oxford University Press outside the submitted work. All other authors declare that they have no competing interests.
Patient consent Not required.
Ethics approval Sydney Children’s Hospital Network Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.