Article Text

Abstract
Objective Evaluate the feasibility and acceptability of routine aspirin in low-risk women, compared with screening-test indicated aspirin for the prevention of pre-eclampsia and fetal growth restriction.
Design Multicentre open-label feasibility randomised controlled trial.
Setting Two tertiary maternity hospitals in Dublin, Ireland.
Participants 546 low-risk nulliparous women completed the study.
Interventions Women underwent computerised randomisation to: Group 1—routine aspirin 75 mg from 11 until 36 weeks; Group 2—no aspirin and; Group 3—aspirin based on the Fetal Medicine Foundation screening test.
Primary and secondary outcome measures (1) Proportion agreeing to participate; (2) compliance with protocol; (3) proportion where first trimester uterine artery Doppler was obtainable and; (4) time taken to issue a screening result. Secondary outcomes included rates of pre-eclampsia and small-for-gestational-age fetuses.
Results 546 were included in the routine aspirin (n=179), no aspirin (n=183) and screen and treat (n=184) groups. 546 of 1054 were approached (51.8%) and enrolled. Average aspirin adherence was 90%. The uterine artery Doppler was obtained in 98.4% (181/184) and the average time to obtain a screening result was 7.6 (0–26) days. Of those taking aspirin, vaginal spotting was greater; n=29 (15.1%), non-aspirin n=28 (7.9%), OR 2.1 (95% CI 1.2 to 3.6). Postpartum haemorrhage >500 mL was also greater; aspirin n=26 (13.5%), no aspirin n=20 (5.6%), OR 2.6 (95% CI 1.4 to 4.8).
Conclusion Low-risk nulliparous women are open to taking aspirin in pregnancy and had high levels of adherence. Aspirin use was associated with greater rates of vaginal bleeding. An appropriately powered randomised controlled trial is now required to address the efficacy and safety of universal low-dose aspirin in low-risk pregnancy compared with a screening approach.
Trial registration number ISRCTN (15191778); Post-results.
- preeclampsia
- screening
- aspirin
- feasibility
- low risk
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Robust multicentre randomised controlled trial design.
Three methods were used to assess aspirin adherence.
Standardisation of methods.
Potential introduction of reporting bias through open-label design.
Introduction
Low-dose aspirin use prior to 16 weeks gestation can reduce the incidence of pre-eclampsia in at-risk pregnancies. When given at this stage, at a dose of 75 mg, the efficacy in low-risk pregnancies is unknown.1 2 With the emergence of first trimester screening tests for pre-eclampsia such as that of the Fetal Medicine Foundation (FMF), one can predict from 11 weeks', the risk of pre-eclampsia.3 Internationally, there are conflicting consensus statements on screening methods and which women meet criteria for aspirin use.4 Application of the FMF screening test and provision of low-dose aspirin to screen positive women can significantly reduce the incidence of early-onset pre-eclampsia (4.3% aspirin vs 1.6% placebo, p=0.004), although predictive performance of the algorithm appears to vary between populations.5 It has been proposed that performance of the FMF algorithm is superior to the methods recommended by the National Institute for Health and Care Excellence (NICE) and American College of Obstetricians and Gynecologists.6 It may be more efficacious to prescribe low-dose aspirin universally, although there is no evidence to support such a policy as yet.7 To determine this, one must first evaluate if low-risk women are willing to take aspirin in pregnancy and if undergoing a comprehensive screening test is realistic in a routine setting. Hence, the primary objective of this multicentre open-label feasibility randomised controlled trial (RCT) was to evaluate the acceptability and feasibility of women taking aspirin 75 mg from beyond 11 weeks’ gestation versus screening-test indicated aspirin. Secondary outcomes included rates of: (1) pre-eclampsia; (2) small-for-gestational-age (SGA) infants; (3) preterm delivery; (4) admission to neonatal intensive care; (5) placental abruption; (6) any reported death and; (7) acceptability of women taking aspirin routinely versus test-indicated aspirin, assessed by a questionnaire.
Methods
Study design
This open-label feasibility multicentre RCT was performed in two Irish tertiary maternity hospitals with 18 000 deliveries per annum collectively. The aim was to include three centres; however, there was a delay in the local ethics committee decision for the third centre (subsequently approved), which was excluded in the interests of study schedule. The protocol for this multicentre RCT has been published8 and was prospectively authorised by the Health Products Regulatory Authority and National Maternity Hospital Central Ethics Committee. The trial was supported by Perinatal Ireland Health Research Board (HRB) and the HRB Mother and Baby Clinical Trials Network following external peer-review for scientific quality. The funder of the study had no role in study design, data collection, data analysis, data interpretation or writing of the manuscript. An independent Trial Steering Committee and Data Monitoring Committee met quarterly to oversee the safety of the trial participants.
Nulliparous women over 18 years old between 11 and 13+6 weeks’ gestation with a viable singleton pregnancy who did not meet criteria for taking aspirin based upon major pre-eclampsia risk-factors (chronic kidney disease; autoimmune disease, eg, systemic lupus erythematosus, diabetes mellitus and chronic hypertension) were eligible for inclusion and thus were recruited at antenatal booking clinics selected at random.9 In Ireland, it is currently not routine obstetric practice to recommend aspirin in women who do not have an aforementioned major risk factor for pre-eclampsia as defined by NICE.9 Exclusion criteria included participants already taking part in a clinical trial, co-existence of a fetal congenital anomaly at recruitment or those with aspirin hypersensitivity. All participants provided written informed consent and were recruited by the research clinician at the first trimester antenatal booking visit.
Randomisation
Participants underwent enrolment and online computerised randomisation by the study sonographer or clinician based on blocks of six to: Group 1—aspirin 75 mg from 11 to 13+6 weeks' once daily until 36 weeks’ gestation; Group 2—no aspirin and; Group 3—aspirin depending on the result of the FMF screening test. Subjects in non-aspirin taking groups underwent routine antenatal care. The randomisation sequence was determined prior to study commencement by the off-site statistician and was concealed from assessors, with both the assessor and participant seeing the group allocation at the same time, following online selection.
Intervention
Enteric-coated Nu-Seals Aspirin (acetylsalicylic acid) 75 mg orally once daily at night from 11 to 36 weeks’ gestation was provided free of charge from Alliance Pharma, which were independent of study protocol and analysis. A dose of 75 mg was used as this is currently the standard recommended dosage in the UK and Ireland for at-risk women.9
Aspirin adherence was assessed subjectively via patient-reported diary cards and tablet counts (checked by both the research clinician and pharmacist) and objectively by assessment of change in urinary 11-dehydroxo-thromboxane-B2 (TxB2). Any reduction in TxB2 between first (pre-aspirin) and second trimester (post-aspirin) levels was taken to suggest that a subject had ingested aspirin within the last 10 days.10
Baseline review and follow-up
Participants underwent two scheduled study visits, at study recruitment and again at 20–22 weeks' (to coincide with their fetal anatomy scan which was performed at the same time) with diary cards and aspirin tablets returned to the research team at 36 weeks’ gestation. Participants completed an anonymous questionnaire at 20–22 weeks based on acceptability of taking aspirin in pregnancy.
Study assessments at the time of the recruitment visit included the FMF screening test, the results of which were assessed for those in Group 3 (screen positive and received aspirin (3A) and screen negative no aspirin (3B)). The FMF screening test was not routine practice within Ireland. Components of the screening test included: maternal history (including ovulation-induced conception, race, body mass index, age, mother with pre-eclampsia); mean arterial blood pressure (MAP); uterine artery Doppler pulsatility index and; pregnancy-associated plasma protein-A (PAPP-A) and placental-like growth factor (PLGF) multiples of the median. To determine the risk of pre-eclampsia, the FMF algorithm was used and based on a screen positive rate of 5%, a cut-off for pre-eclampsia prior to 42 weeks at greater than 1:8 was used.3 This cut-off was selected with the aim of capturing the majority of pre-eclamptics; both preterm and post-term and at the time of study commencement, this was the optimal screening algorithm for the detection of any pre-eclampsia. Two non-blinded trained clinical research sonographers acquired the first trimester uterine artery Doppler waveforms and MAP and interpreted the findings. MAP was assessed using an automated blood pressure monitoring device as outlined by the technique stipulated by the FMF.11 Uterine artery Doppler velocimetry was acquired using Viewpoint Version 5.6.16 GE Healthcare, 2012 and Voluson Expert 730, GE 2012 using the technique outlined by the International Society of Ultrasound in Obstetrics and Gynecology. The pulsatility index was measured from both uterine arteries and an average value was calculated.12
A maternal blood sample was analysed for PAPP-A and PLGF under standard conditions using a 6000 DELFIA Xpress, PerkinElmer, 2014 clinical random access screening platform in the hospital clinical biochemistry laboratory. A quantitative immunoturbimetric TxBCardio immunoassay was used to determine TxB2 levels in urine samples obtained via both study visits. These were then standardised as a ratio with creatinine levels and expressed as pg/mg creatinine.
Outcomes
The primary objective of this study was to evaluate the feasibility and acceptability of low-risk nulliparous women taking aspirin versus test-indicated aspirin in pregnancy. Outcome measures included:
The proportion of eligible women agreeing to participate in a trial where aspirin is prescribed routinely (feasibility).
Compliance with study protocol, as measured by the following: (1) adherence to aspirin (acceptability), (2) attendance at study visits (acceptability), (3) satisfactory collection of all endpoints and variables (feasibility), (4) specific study protocol violations (feasibility).
The proportion of women in whom it was possible to obtain first trimester transabdominal uterine artery Doppler velocimetry examination (feasibility).
Proportion of women with a completed screening test who were issued the screening result within 1 week of having the test performed (feasibility).
Secondary outcomes included rates of: (1) pre-eclampsia; (2) SGA infants (customised sex-specific birth weight <10th centile); (3) preterm delivery prior to 34 weeks; (4) admission to the neonatal intensive care unit (NICU); (5) placental abruption; (6) any reported death (stillbirth, neonatal or infant death) and (7) acceptability of women taking aspirin routinely versus test-indicated aspirin as assessed by an anonymous questionnaire at 20–22 weeks. As part of routine antenatal care, women had an appointment with their midwife and/or clinician at booking (11–14 weeks'), 16 weeks', 18–20 weeks', 25 weeks', 28 weeks', 31 weeks', 34 weeks', 36 weeks', 38 weeks', 40 weeks' and 41 weeks' of gestation in line with hospital protocol. During each visit, blood pressure was assessed using mercury sphygmomanometry and a urine dipstick for proteinuria was performed with symphysis-fundal height and or fetal biometrical ultrasound assessment as appropriate. Pre-eclampsia was defined based on the definition from the International Society for the Study of Hypertension in Pregnancy, with new-onset hypertension (>140 mm Hg systolic or >90 mm Hg diastolic) after 20 weeks’ gestation associated with: (1) proteinuria of at least 1 g/L (2+) on urine dipstick testing, (2) maternal organ dysfunction and/or (3) fetal growth restriction (FGR).13 Suspicion of a diagnosis of pre-eclampsia at an antenatal visit prompted further investigation in the fetal assessment unit with clinical examination, blood testing (urea and creatinine, liver function tests and full blood picture), 24-hour urine collection for proteinuria and departmental fetal ultrasound assessment with final diagnosis made by an obstetrician.
Safety data were reported as adverse and serious adverse events and participants that discontinued from the study were recorded in addition to the reason for discontinuation and outcome. As an assessment of postpartum haemorrhage, blood loss was weighed at the time of delivery.
Statistical analysis
As outlined in the published study protocol, the projected sample size for this study was 500 women across two sites with 18 000 deliveries per annum collectively.8 To determine pre-eclampsia as a primary outcome, the anticipated number of patients required is over 15 000 women. As this study aimed to determine the feasibility of such a study, 500 participants were more than adequate as 3% of the number required for a substantive study is required (n=450).14 Accounting for a dropout rate of 10% (n=45), 500 participants were adequate to obtain the primary outcome. Analysis was performed by a statistician using SAS v.20 on the intention-to-treat (ITT) population, which included all participants randomised, who had completed the full second trimester assessment. Measures of variance included SD. Follow-up of serious adverse events continued until 28-days following delivery. Adverse events were reported as odds ratios (ORs) and uncertainty was expressed using 95% CIs. No hypothesis tests were performed.
Patient involvement
Although patients were not directly involved in devising the study protocol and design, the burden of the RCT intervention (ie, taking aspirin and undergoing the FMF screening test) was assessed by means of an anonymous questionnaire completed at 20–22 weeks’ gestation. At the time of study participation, subjects were informed that study results could be viewed following publication on the study website http://perinatalireland.ie/research/test/
Results
Subjects were recruited between the 8 May 2014 and 23 September 2015 with follow-up of participants until 11 April 2016, when the study was ended by the steering committee following delivery of the final patient as the target sample size had been achieved. In total, 1054 eligible women were approached to take part in the study and of these, 557 underwent randomisation (figure 1). In the screen and treat population (Group 3) n=184, 13 (7.1%) women had a risk of developing pre-eclampsia >1:8 and subsequently started taking aspirin until 36 weeks’ gestation. Eleven women were excluded from the study leaving 546 in the ITT population. In total, there were 192 women in the ITT group who were taking aspirin as per randomisation and 354 not taking aspirin. Baseline characteristics were similar and the summaries are presented in table 1.
Baseline characteristics of the study population

Consort diagram.
Primary outcomes
The proportion of eligible women agreeing to participate in a trial where aspirin is prescribed routinely (feasibility); 1054 women were approached who were eligible to partake. 497 were subsequently not enrolled as they did not want to take aspirin n=454 or for an alternative reason n=43, for example, appointment did not suit. Hence, 546/1054 (51.8%) women were willing to partake in a study where they may have to take aspirin routinely.
Compliance with study protocol, as measured by the following: (A) adherence to aspirin (acceptability), (B) attendance at study visits (acceptability), (C) satisfactory collection of all endpoints and variables (feasibility), (D) specific study protocol violations (feasibility):
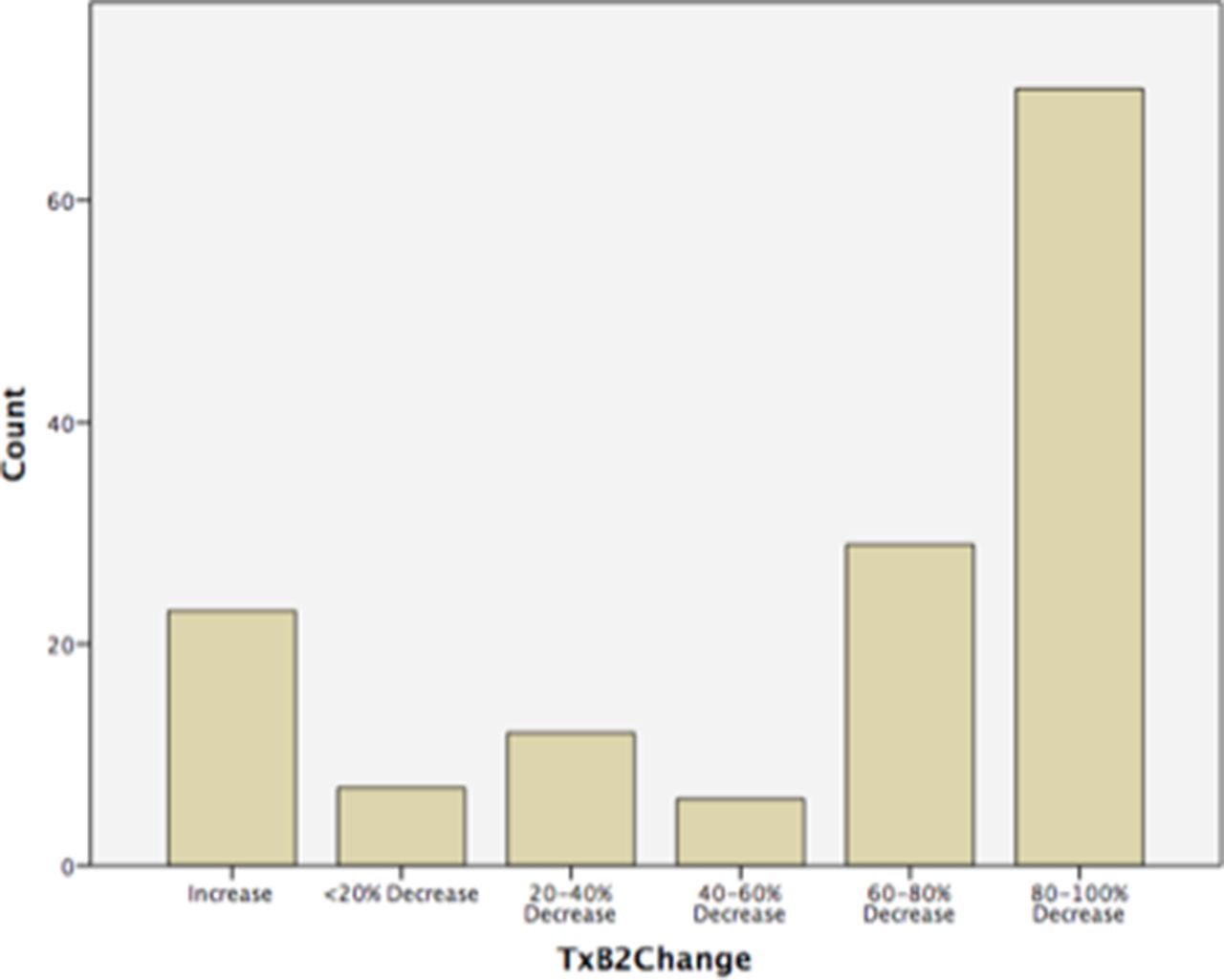
Of those women included in the analysis who were taking aspirin (n=192), the average adherence based on patient-reported diary cards was 96.0% and based on tablet counts it was 95.0%. Seven women were non-adherent and 19 (9.9%) were poorly compliant (<80%). Average adherence was 95.0% in both the test-indicated aspirin group (3A) and routine aspirin group (1) (table 2). The median first trimester pre-aspirin urine TxB2 level was 8662.2 pg/mg (IQR 2014.5–9931.5) and second trimester (post-aspirin) 2285.1 pg/mg (IQR 591.0–2300.1). The percentage change in TxB2 was then assessed for all paired samples (n=147) and found that 124/147 (84.4%) of subjects had a fall in TxB2 levels between the first and second trimesters versus 23/147 (15.6%) who had an increase. The greater the reduction in urinary TxB2 pre-aspirin and post-aspirin dose, the greater the degree of aspirin adherence, as demonstrated in figure 2. Patient groups were similar (routine aspirin and screen-positive aspirin) in relation to adherence measures of tablet counts, diary cards and percentage change in urine TxB2.
Of those who underwent randomisation (n=557), 11 were excluded prior to fulfilment of study participation requirements (attendance at second study visit). Of the 11, three withdrew consent for participation as they decided that they did not wish to take aspirin following randomisation.
Of all 546 subjects, collection of outcome measures and variables were obtained for all apart from the questionnaire on patient acceptability, which was completed in 97.1% (530/546).
Six protocol violations were recorded (0.01 per 100 participants) including women transferring care to another hospital (n=3), incorrect randomisation of women who did not meet inclusion criteria (n=2) and a subject in the non-aspirin group who started taking aspirin on the advice of their clinician (n=1).
The proportion of women in whom it was possible to obtain first trimester transabdominal uterine artery Doppler velocimetry (feasibility); the FMF screening test was completed in 98.4% (181/184) following successful uterine artery Doppler velocimetry acquisition, of which one was obtained vaginally due to challenges with abdominal acquisition, with an overall sonographer reported ease of acquisition 3.1 (SD ±0.91) (score 1 (easy) to 5 (unobtainable)) (table 2).
The proportion of women with a completed screening test issued the result within one week of the test (feasibility); the average time to obtain the laboratory analysed PAPP-A and PLGF results so that a screening result could be issued was 7.6 days (0–26) with 78 (42.4%) of women waiting greater than one week and five women being beyond 16 weeks' prior to result availability (table 2).

{kind=link}
{kind=link}
Histogram demonstrating percentage change in urinary thromboxane-B2 levels pre-aspirin and post-aspirin administration (n=147) (TxB2=urinary thromboxane level).
Primary outcomes of feasibility and adherence
Secondary outcomes
There was no difference between groups in relation to secondary outcomes (online supplementary table S1). For the overall cohort, there were three cases of early-onset pre-eclampsia <34 weeks (0.55%), n=22 (4.03%) any pre-eclampsia, n=57 (10.44%) SGA infants and 15.02% (n=82) placental disease. Secondary outcomes for groups 3A (screen-positive aspirin) and 3B (screen-negative no aspirin) are demonstrated in online supplementary table S2. Despite taking aspirin, there remained a greater number with pre-eclampsia at <37 weeks in the screen-positive versus the screen-negative group, although numbers were small (n=2 (15.4%) vs n=2 (1.2%)). In terms of taking aspirin in a subsequent pregnancy, the questionnaire revealed that 92.3% (489/530) were willing to take aspirin in a subsequent pregnancy; 92.5% (173/187) of aspirin takers and 91.5% (314/343) of non-aspirin takers.
Supplementary file 1
Supplementary file 2
Safety
The adverse event profile differed between groups but not the serious adverse event profile (table 3 and online supplementary table S3). There were six perinatal deaths, all of which underwent postmortem. In the aspirin group, there was one placental abruption and one case of intervillous haemorrhage. Perinatal deaths in the non-aspirin groups were due to delayed villous maturation, severe FGR, fetal thrombotic vasculopathy and neonatal septicemia. There was an observable difference between groups in terms of reported vaginal spotting aspirin 15.1% versus non-aspirin 7.9% OR 2.1 (CI 1.2 to 3.6), which was not associated with pregnancy loss. Similarly, the rate of PPH >1000 mL was higher in the aspirin group. However, the numbers were small. Rates of blood transfusion or significant haemoglobin drop to <8 g/dL were similar.
Supplementary file 3
Adverse and serious adverse events in aspirin and non-aspirin taking groups
Discussion
Main findings
This feasibility RCT has found that low-risk nulliparous women were open to taking aspirin in pregnancy and were adherent, with a willingness to take it again in a subsequent pregnancy. We can say this as, comparing findings to other RCTs in pregnancy, of which there are few, the uptake in this RCT was much higher, as was adherence (eg, Chiswick, et al 2015; 35% enrolment and 65%–67% adherence with metformin use).15 This is the first trial of its kind, which has assessed the acceptability of women taking aspirin in low-risk pregnancy and the feasibility of an integrated screening test in a routine clinical setting.
Strengths and limitations
The strengths of this study are the multicentre RCT design with robust protocol and oversight and previously published methodology. Allocation bias was limited by the use of a prospective approach and selection bias was limited through randomisation. The fact that the same two sonographers and biochemists were responsible for conducting the screening test with the use of quality control standards for test completion using the same equipment and technique for all subjects optimised reproducibility. There were a low number of dropouts and almost all patient outcomes were recorded. Although there is currently no validated scientific method of assessing aspirin adherence,16 a laboratory assessment of change in TxB2 served as a more objective measure, thus strengthening reliability. Study weaknesses were primarily that PAPP-A and PLGF analysis was performed in the laboratory using validated methods with quality assurance, as opposed to the bedside point-of-care tests, hence it took longer to obtain a result. In a non-research setting with a greater throughput of patients, one could anticipate a faster turnaround time. Additionally. the open-label nature of the study meant that safety recording was open to reporting bias, and as is often the case with RCTs the uptake of subjects demonstrated dominance for educated women. In RCTs, there is always a risk of introducing a Hawthorne effect, whereby subjects act differently in the confines of an RCT as to how they would in a real-life setting, hence adherence rates may have been over-represented.17 A third trimester visit may have added strength to the study to assess objectively for aspirin adherence and patient satisfaction; however, as adherence prior to 16 weeks was deemed the critical time point for pre-eclampsia prevention, follow-up at 20–22 weeks was selected.
Interpretation
A recently published large RCT from the FMF found that, following application of FMF screening and subsequent randomisation of women deemed to be at risk of preterm pre-eclampsia to aspirin 150 mg versus placebo, there was a reduction in the incidence of preterm pre-eclampsia in the aspirin arm.5 Our study differs on several counts: (1) routine aspirin arm—use of a third arm assessing provision of routine aspirin assessed the acceptability and feasibility of this policy; (2) aspirin dosage (150 mg vs 75 mg)—in light of limited evidence on dosage and effect, the safest lowest effective dose was selected. A recent meta-analysis, published since completion of this study suggests that there is an aspirin dose–response effect, with higher doses of aspirin taken prior to 16 weeks’ gestation, associated with a greater reduction in pre-eclampsia and FGR compared with standard lower doses.18 When supported by robust safety data when using higher dosing, this is something to consider in future studies and clinical practice; (3) adverse events—rates of PPH and vaginal bleeding were reported. This information would be useful from the FMF study in light of the higher aspirin dosing regimen and; (4) our study was not powered to detect a difference in clinical outcome, with the primary focus being feasibility and acceptability.
Few studies have assessed the acceptability of non-routine medications in pregnancy. In the developing world, pregnant women are willing to take calcium, oral iron and micronutrients.19–21 If instructed about potential side-effects and reminded frequently, women had higher levels of adherence with the greatest barrier being forgetfulness. Average medication adherence in pregnancy for chronic illness is higher than for non-routine medications at 90%–95%,22 hence it its promising that we have noted a rate as high as this in our own study. There was a slight discrepancy in adherence assessed via tablet counts and diary cards and that more objectively assessed via TxB2. Reasons for this may include the potential for aspirin resistance, which although not formally assessed in this study can be increased when using an enteric-coated preparation.23
The FMF screening test was feasible in terms of acquiring first trimester uterine artery Doppler velocimetry measurements, though delays were encountered in obtaining laboratory analysed PAPP-A and PLGF. This is relevant as it reflects the practical aspects of such a screening test in a clinical real-life setting. Improved protocols between the clinical and laboratory staff would be required to allow patients receive results within a reasonable timeline.
In terms of vaginal spotting and clinically significant PPH with aspirin use, the findings of this study are comparable with previous studies although evidence of increased antenatal and postnatal bleeding requires further investigation, most notably with the use of aspirin at doses greater than 75 mg.24 25 Due to the open-label nature of this study as opposed to placebo control, there is always a potential of reporting bias of bleeding in the aspirin arms. Although generally safe in pregnancy, it may be worthwhile considering cessation of aspirin at 32–34 weeks’ gestation with the aim of reducing the risk of PPH, as opposed to 36 weeks' and of informing women of the unwanted side effect of increased vaginal spotting.
Conclusion
It has been proposed that the most cost-effective approach to reducing pre-eclampsia is the provision of an effective, affordable and safe intervention applied to all mothers without prior testing to assess levels of risk.7 An algorithm-based screen-and-treat approach, as proposed by the FMF can reduce rates of preterm pre-eclampsia when doses of 150 mg of aspirin are used. This study was not powered to detect a difference in rates of pre-eclampsia between groups, yet has taken the first step to address if low-risk nulliparous women are open to taking aspirin in the first instance and if application of a screening algorithm is feasible. Moving forward, an RCT is now required to address the efficacy of universal low-dose aspirin in low-risk pregnancy compared with a screening-based approach. This will require significant numbers due to the low incidence of early-onset pre-eclampsia. Although women were open to taking aspirin in pregnancy compared with other RCTs involving medication, almost twice the number enrolled had to be approached to obtain adequate study participants. This must be considered when planning a future trial.
Acknowledgments
The authors acknowledge women who took part in the study.
References
Footnotes
Contributors Conceived and designed the experiments: FM, CM, PMP, FB, PDo, DMC, MC, AS, FC, JM, SD, JH, AC, ECT, PDi, ZA, FDM, FMA. Performed the experiments: FM, CM, FC. Analysed the data: FM, PD, ZA, FMA. Contributed reagents/materials/analysis/tools: PMP, FB, PDo, DMC, MC, AS, JM, SD, JH, AC, AH, ECT, PDi, ZA, FDM, FMA. Wrote the paper: FM, CM, PMP, FB, PDo, DMC, MC, AS, FC, JM, SD, JH, AC, AH, ECT, PDi, ZA, FDM, FMA. All coauthors approved changes to the manuscript following reviewer comments.
Funding This work was supported by Perinatal Ireland HRB and HRB Mother and Baby Clinical Trials Network.
Competing interests None declared.
Patient consent Not required.
Ethics approval National Maternity Hospital Central Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Dataset available from corresponding author on request.