Article Text

Abstract
Introduction Hereditary proximal spinal muscular atrophy (SMA) is caused by homozygous loss of function of the survival motor neuron 1 gene. The main characteristic of SMA is degeneration of alpha motor neurons in the anterior horn of the spinal cord, but recent studies in animal models and patients have shown additional anatomical abnormalities and dysfunction of the neuromuscular junction (NMJ). NMJ dysfunction could contribute to symptoms of weakness and fatigability in patients with SMA. We hypothesise that pyridostigmine, an acetylcholinesterase inhibitor that improves neuromuscular transmission, could improve NMJ function and thereby muscle strength and fatigability in patients with SMA.
Methods and analysis We designed a monocentre, placebo-controlled, double-blind cross-over trial with pyridostigmine and placebo to investigate the effect and efficacy of pyridostigmine on muscle strength and fatigability in patients with genetically confirmed SMA. We aim to include 45 patients with SMA types 2–4, aged 12 years and older in the Netherlands. Participants receive 8 weeks of treatment with pyridostigmine and 8 weeks of treatment with placebo in a random order separated by a washout period of 1 week. Treatment allocation is double blinded. Treatment dose will gradually be increased from 2 mg/kg/day to the maximum dose of 6 mg/kg/day in four daily doses, in the first week of each treatment period. The primary outcome measures are a change in the Motor Function Measure and repeated nine-hole peg test before and after treatment. Secondary outcome measures are changes in recently developed endurance tests, that is, the endurance shuttle nine-hole peg test, the endurance shuttle box and block test and the endurance shuttle walk test, muscle strength, level of daily functioning, quality of and activity in life, perceived fatigue and fatigability, presence of decrement on repetitive nerve stimulation and adverse events.
Ethics and dissemination The protocol is approved by the local medical ethical review committee at the University Medical Center Utrecht and by the national Central Committee on Research Involving Human Subjects. Findings will be shared with the academic and medical community, funding and patient organisations in order to contribute to optimisation of medical care and quality of life for patients with SMA.
Trial registration number NCT02941328.
- spinal muscular atrophy
- sma
- neuromuscular junction
- pyridostigmine
- fatigability
- cross-over
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This randomised double-blind, placebo-controlled cross-over trial will provide important information to clinicians and patients with spinal muscular atrophy (SMA) about efficacy of treatment of fatigability, lack of endurance and diminished motor function with pyridostigmine.
The cross-over design is an ideal design for this rare disease with striking variability because participants will be their own controls which reduces unsystematic variance, subsequently reducing the necessary sample size to detect systematic variance after treatment.
Permuted block randomisation ensures treatment group numbers are evenly balanced at the end of each block and at the end of the study with this relatively small number of participants.
The use of tests that are still in the process of validation is a limitation of this protocol. However, these tests can capture a dimension of SMA for which validated outcome measures are largely lacking.
Introduction
Hereditary proximal spinal muscular atrophy (SMA) is a motor neuron disease in children and adults caused by a homozygous deletion of the survival motor neuron 1 (SMN1) gene or a heterozygous deletion combined with a loss-of-function mutation on the other allele, resulting in a significant reduction of full length functional SMN protein.1 2 The main characteristic of SMA is the degeneration of alpha motor neurons in the anterior horns of the spinal cord, resulting in progressive muscle weakness of axial muscles and muscles of the arms and legs with a mild to severely reduced life expectancy in the majority of patients.3–5
SMN protein is ubiquitously expressed and is involved in the pre-mRNA splicing pathway, ubiquitin and cytoskeleton homoeostasis, endocytosis and axonal transport.6–10 Although motor neurons are most sensitive to the disruption of cellular pathways caused by SMN deficiency, other cell types and tissues may be affected as well.11 12 Histological and electrophysiological studies have shown that sufficient levels of SMN protein are essential for the development, maturation and function of the neuromuscular junction (NMJ).13 14 SMN-deficient mice display both presynaptic (ie, abnormal density and distribution of synaptic vesicles and abnormal accumulation of neurofilaments at the nerve terminal of the NMJ) and postsynaptic (ie, shrinkage of motor endplates) abnormalities.15–18 In patients with SMA type 1, abnormal aggregation of acetylcholine receptors (AChRs) at the muscle endplates has been reported.18 19 Nerve conduction studies with repetitive nerve stimulation (NCS-RNS) in patients with SMA types 2 and 3, a specific but not very sensitive test for NMJ dysfunction, showed an abnormal decremental response in 49% of patients.13 Patients with SMA frequently complain of fatigability, which is defined as a decrease in performance over a given time or sustained measure of mechanical output,20 in addition to muscle weakness.
Intrathecal administration of SMN-specific antisense oligonucleotides that augment cellular SMN levels improves motor development in infants and children with SMA, but efficacy has not been tested in adults.21 22 There is a clear need for low-cost treatment that is easy to administer in patients with longer disease duration. The finding of postsynaptic dysfunction of the NMJ in SMA suggests that patients may benefit from drugs that facilitate neuromuscular transmission. Acetylcholinesterase inhibitors may represent a new category of candidate drugs for the treatment of SMA. Pyridostigmine, an acetylcholinesterase inhibitor with relatively long half-life, is an FDA and EMA approved first-line treatment of disorders of the postsynaptic NMJ, that is, myasthenia gravis. Pyridostigmine inhibits the natural enzymatic breakdown of acetylcholine and thereby increases its biological availability at the NMJ enhancing neuromuscular transmission.23
Our aim in this study is to investigate the efficacy and effect of pyridostigmine on muscle strength and fatigability in SMA. We designed a placebo-controlled, cross-over trial in patients with SMA types 2, 3 or 4, with double-blind treatment allocation. The cross-over design is an ideal design for this rare disease with striking variability, as using participants as their own controls will reduce the unsystematic variance (error variance). This allows for easier detection of systematic variance following the intervention using fewer study participants. The short half-life of pyridostigmine minimises carry-over effects.
Methods and design
Study setting and design
We conduct this study at the neuromuscular department of the University Medical Center Utrecht, a tertiary referral centre for neuromuscular diseases in the Netherlands. All members of the study team, consisting of physicians, physical therapists and nurses, have broad experience with SMA due to the national cohort study that is carried out in this centre since 2010.24
This investigator-initiated, monocentre, placebo-controlled study has a cross-over, double-blinded design, with blinding of participants and investigators. The pharmacist is not blinded for allocation of treatment. The study protocol was designed using the recommendations of the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines. (See online supplementary additional file 1 for the SPIRIT checklist 2013 statement.)
Supplementary file 1
The study is currently ongoing; the first participant was included on 25 November 2015. We expect study completion by the end of 2017.
Participants
The details of the inclusion and exclusion criteria are provided in box 1. The main inclusion criteria are: a clinical diagnosis of SMA types 2, 3 or 4 and a genetically confirmed homozygous SMN1 deletion and age 12 years or older. We recruit patients with SMA types 2–4 through the national SMA registry that contains detailed clinical information of approximately 300 patients.24 To minimise selection bias, all eligible patients, based on known SMA type, are invited to participate.
Selection criteria
Inclusion criteria
Clinical diagnosis of SMA types 2, 3 or 4
Type 2: age at onset >6 months and ability to sit unsupported but not to walk unsupported.
Type 3: age at onset >18 months and the ability to walk unsupported at any point in life.
Type 4: age at onset ≥30 years and the ability to walk unsupported at any point in life.
In case of discrepancy between age at onset and highest acquired motor milestone, the latter is used to define SMA type.
Genetically confirmed homozygous SMN1 deletion.
Given oral and written informed consent when ≥18 years old and additional informed consent by the parents or legal representative in case of participants aged ≥12 till <18 years old.
Ability to perform at least two subsequent rounds of the nine-hole peg test.
A maximum Motor Function Measure score of 80%.
Exclusion criteria
Known concomitant disorders of the NMJ (Lambert-Eaton myasthenic syndrome, myasthenia gravis).
Use of drugs that may alter NMJ function
Cholinergic medication (eg, rivastigmine, neostigmine, galantamine, physostigmine, succinylcholine).
Non-depolarising muskelrelaxans (eg, (cis)atracurium, gallamine, mivacurium, pancuronium, rocuronium, vecuronium).
Other antagonising medication of pyridostigmine (procainamide, quinidine, propranolol, lithium, chloroquine, hydroxychloroquine , aminoglycoside antibiotics, clindamycin, polymyxin).
SMA type 1.
Apprehension for participation in nerve conduction studies.
Inability to meet study visits.
Mechanical gastrointestinal, urinary or biliary obstruction.
Clinical significant alterations of blood tests drawn within 14 days prior to start of study entry.
Electrocardiophysiology abnormalities known as a contraindication for pyridostigmine use.
Current pregnancy or breast feeding.
Known allergy to bromides.
Severe bronchial asthma (in case of uncertainty of diagnosis, we will contact the treating pulmonologist or physician).
SMA, spinal muscular atrophy; SMN1, survival motor neuron 1; NMJ, neuromuscular junction.
We will not replace withdrawn or unblinded participants. We will not include them in the study again once dropped out, and we will not reuse their identification number and treatment. With permission of the participant, we will plan a follow-up by phone for at least 1 week. If there is an adverse event (AE) that is still present after 1 week, follow-up will be longer.
Sample size calculation
We aim to recruit 45 participants with SMA types 2–4 based on two power calculations that we performed based on the cross-over design using pilot data on repeated measures of the total score of the Motor Function Measure (MFM) test (unpublished data). First, we calculated the within-participant SD, and next the SD of the difference between subsequent measurements of the participants.
Calculation 1: If a total of 40 participants will enter this two-treatment cross-over study, the probability is 80% that the study will detect a treatment difference at a two-sided 0.05 significance level, if the true difference between treatments is 1.093 units. This is based on the assumption that the within-participant SD of the response variable is 1.7 units.
Calculation 2: If a total of 40 participants will enter this two-treatment cross-over study, the probability is 80% that the study will detect a treatment difference at a two-sided 0.05 significance level, if the true difference between treatments is 1.409 units. This is based on the assumption that the SD of the difference in the response variables is 3.1 units.
Both calculations show similar results in terms of the detectable difference based on 80% power, a two-sided significance level of 0.05 and 40 participants in total in the trial. Five additional participants will be recruited to compensate for potential drop-outs. The total number of included participants will therefore be 45.
Intervention and participant timeline
Once the investigator confirms eligibility, the participant is assigned to one of the two treatment groups (pyridostigmine or placebo) and will crossover during the trial.
Randomisation to a treatment order (ie, to start with either pyridostigmine or placebo) is done through a permuted four-block design by the pharmacist, who is the only one not blinded for treatment allocation. Permuted block randomisation ensures treatment group numbers are evenly balanced at the end of each block and at the end of the study with this relatively small number of participants.
Figure 1 shows the participant timeline. At the screening visit, we investigate whether participants are eligible for participation in the trial concerning all inclusion and exclusion criteria. As a safety measure, participants are screened for clinical significant alterations in blood tests (sodium, potassium, haemoglobin, haematocrit, C reactive protein, urea, creatinine, estimated glomerular filtration rate, aspartate transaminase, alanine transaminase, gamma-glutamyl transpeptidase, anti-AChR antibodies and beta-human chorionic gonadotropin (HCG) levels as a pregnancy test), and they are screened for ECG alterations to ensure that included participants have no kidney dysfunction, liver function alterations, bradycardia or arrhythmias and/or a present pregnancy. If any of the screening tests results in a clinically significant alteration, the participant is excluded from study participation. The study schedule is presented in table 1. At the start of the study, participants are randomised to one of two intervention groups (double blinded; A or B). Each participant receives pyridostigmine (tablet) and placebo (matching tablet with no pharmacological ingredients) in consecutive periods.

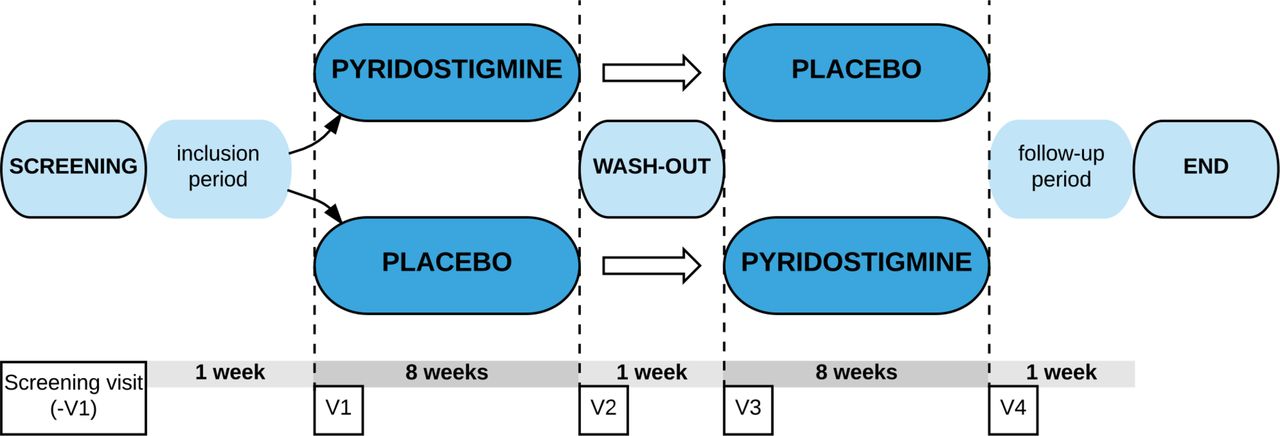{kind=link}
Flow chart of the study protocol visit 1 has to take place 5–14 days after the baseline visit. Visit 2 has to take place 7–9 weeks after visit 1. The washout period consists of at least 7 days up to a maximum of 14 days. Visit 3 is planned at the end of the washout period. Visit 4 has to take place 7–9 weeks after visit 3. There is no physical close out visit. Participants are instructed to contact the study team if any events occur in the first week after last intake of study medication. V, visit.
Trial schedule of enrolment, interventions and assessments
Group A starts with 8 weeks of treatment with pyridostigmine at a final dose of 6 mg/kg/day. After a 1-week washout, they start an 8-week period with placebo treatment.
Group B starts with 8 weeks of treatment with placebo. After a 1-week washout, they start an 8-week period with pyridostigmine at a final dose of 6 mg/kg treatment per day.
Each treatment is given four times per day and dosage is gradually increased in the first week of each treatment period to minimalise side effects; starting at 2 mg/kg/day in the first 3 days after the first administration. When this dose is well tolerated, the dose is increased to 4 mg/kg/day during day 4 up till day 7. When this dose is well tolerated, the dose is increased to the maximum dose of 6 mg/kg/day after 1 week. In case of unfavourable side effects of the medicinal product at 6 mg/kg/day, the participant continues to use the highest achievable dose (2 or 4 mg/kg/day). The investigator gives the approval for increase of the dosage after the first 3 days and after 7 days by phone, after checking for invalidating side effects. If there are side effects, the investigator can decide to not increase the dosage or to (temporarily) stop the medication depending on the extent of the side effects. In case of severe side effects, the investigator can decide to intervene. Pyridostigmine can cause a cholinergic crisis when overdosed due to the parasympathomimetic induction. Symptoms of a cholinergic crisis are excessive salivation, urinary urgency, diarrhoea, muscle weakness, fasciculations, cramps of striated muscles and respiratory problems. In case of symptoms of diarrhoea, excessive salivation and or sweating atropine sulfate can be given orally, 0.125 mg one to two times per day. In case of severe symptoms, these symptoms can be treated with intravenous 1–2 mg atropine sulfate on slow infusion and supportive care of respiratory function, if needed. When it is necessary to unblind the treatment of a participant, for example, in medical emergencies, this is done by the on-call pharmacist. The rest of the study team remains blinded.
To monitor therapy adherence, we inquire participants about any problems taking the medication and we manually count residual study medication and compare this to the expected amount based on their individual treatment schedule.
Prohibited concomitant medication can be found in the exclusion criteria. We ask participants to contact us before starting (prescribed) medication, vitamins or supplements during the study to check for compatibility and to register this change in medication. We also register other events that may influence fatigability (eg, changes in work or school schedules).
Outcome measures
This study investigates the effect and efficacy of pyridostigmine on motor function and fatigability in patients with SMA. The test battery is performed in the same order, at all five visits.
Primary endpoint
Is the change in motor function and fatigability using the following measures:
Motor function and fatigability
MFM: The MFM is a quantitative scale allowing to measure the functional motor abilities of an individual affected by a neuromuscular disease, regardless of the diagnosis and the extent of motor deficiencies. The MFM has been validated in patients with neuromuscular disorders, aged 6–60 years, including patients with SMA. The MFM contains three domains reflecting distal motor function, axial/proximal motor function and total muscle function. Studies in patients with SMA show striking differences in subscores and total scores between patients with different SMA types.25 26 We use the validated English version of the MFM.
Repeated nine-hole peg test (r9HPT): The r9HPT is a modification of the 9HPT targeting endurance instead of motor function. The 9HPT is a brief, standardised, quantitative test of upper extremity function.27 28 The participant is seated at a table with a plastic block containing a small, shallow container holding nine pegs and nine empty holes. On a start command when a stopwatch is started, the participant picks up the nine pegs one at a time, puts them in the nine holes as quickly as possible, and, once they are in the holes, removes them again as quickly as possible one at a time, replacing them into the shallow container. The time to complete the task is recorded. Participants will perform five consecutive rounds with the same hand of choice with the Rolyan 9HPT (Patterson Medical, Homecraft Rolyan; Sutton-in-Ashfield, UK). The score for the 9HPT is an average of the five rounds. We will also look at the change in score per round, suspecting an increase in time needed to perform the test in consecutive rounds when participants do not use pyridostigmine, as a result of the muscle fatigability.
Secondary endpoints
To additionally investigate the effect of treatment on muscle strength and daily life functioning the following measures are used:
Motor function and fatigability
Medical Research Council Scale (MRC scale): The MRC scale is widely accepted and frequently used in the neurology and rehabilitation practice to objectively validate and follow-up on muscle strength.29 30 The MRC scale has successfully been used in multiple trials with SMA types 2–4.31–33 The participant’s effort is graded on a scale of 0–5 (grade 0=no movement observed, grade 5=muscle contracts normally against full resistance). MRC scores of a total of 22 different muscles of both upper and lower extremities are determined.
Endurance Tests: Recently, we developed a panel of endurance tests to assess fatigability/endurance in patients with SMA with a wide range of disease severity, that is, the endurance shuttle nine-hole peg test (ESNHPT), the endurance shuttle box and block test (ESBBT) and a modified version of the endurance shuttle walk test (ESWT). The methodology is based on the original ESWT in which participants have to walk on a predetermined walking speed during a maximal time period of 20 min.34–36 The same methodology is applied to the BBT37 and the NHPT,27 28 creating two endurance tests for the upper extremity. Reliability and validity are being studied in parallel with this study (Bartels et al, in progress). Ambulatory participants perform the ESWT and the ESBBT and non-ambulatory participants perform the ESNHPT and, if possible, the ESBBT, which requires more strength of the proximal arm muscles. Primary outcome measures of these tests are time to limitation and walking distance for the ESWT or number of blocks or pegs for the ESBBT and ESNHPT. We measure maximum isometric strength of five arm muscles and six leg muscles before and directly after the test to determine exercise-induced muscle weakness. Surface EMG is assessed during the endurance test to determine local fatigability response of the muscle. We use the OMNI scale38 39 prior and directly after completion of the test to evaluate perceived exertion.
Patient-reported outcome measures: quality of life
The 36-Item Short Form Health Survey (SF-36): The SF-36 is a standardised, generic health-related quality of life measure in motor neuron40–42 and other diseases.43 The SF-36 covers eight dimensions (physical functioning, role limitations due to physical health problems, bodily pain, generic health perceptions, vitality, social functioning, role limitations due to emotional problems and mental health). The validated Dutch version of the SF-36 is used.43
Paediatric Quality of Life inventory (PedsQL): The PedsQL M 3.0 Neuromuscular Module has been developed in the last decade to measure quality of life dimensions specific to children aged 2–18 years with neuromuscular disorders, in particular, Duchenne and SMA.44–46 The PedsQL encompasses three domains: items on disease process and associated symptomatology, items related to the patient’s ability to communicate with healthcare providers and others about his/her illness and items related to family financial and social support systems.
Patient-reported outcome measures: perceived daily functioning, fatigue and fatigability
SMA-Functional Rating Scale (SMA-FRS): The SMA-FRS is a functional scale modified from the amyotrophic lateral sclerosis functional rating scale (ALSFRS) and the WeeFim protocol.47 48 It reflects important aspects of daily functioning.
Perceptions of Fatigue: In participants, aged 12–17 years, fatigue is assessed with the PedsQL Multidimensional Fatigue Scale.49–51 In participants, aged ≥18 years, fatigue is assessed with the Fatigue Severity Scale.52
Fatigability Questionnaire: Perceived fatigability during activities of daily life is assessed with a questionnaire in all participants. We use the fatigability questionnaire developed for patients with peripheral nerve disorders by Straver et al for adult participants and an adjusted form combined with the Child Health Assessment Questionnaire for children.53 54
Nerve conduction studies
NCS-RNS: Four different muscles are tested (musculus abductor digiti minimi, musculus flexor carpi radialis, musculus trapezius and musculus nasalis) for supramaximal compound muscle action potential (CMAP) recording and 3 Hz repetitive stimulation (train of 10) in rest and after 60 s of maximal voluntary muscle activation.13
Adverse events
All AEs that are reported spontaneously by the participant or observed by the investigator or study staff members are recorded and if necessary, appropriate measures are taken.
Statistical analysis
We will analyse differences in baseline characteristics between participants for single measures (ie, age, disease duration) using t-tests or non-parametric tests, depending on the distribution of data. We will use a linear mixed-effects model to analyse differences in outcome for the different treatment arms. Treatment arms will be entered as fixed effect, while the repeated measurements on the participants will be entered as random effects. A linear mixed-effects model for repeated measures allows us to additionally adjust for age, disease duration, gender, SMA type and other possible influencing factors. We primarily focus on the results in the group as a whole, but we will additionally stratify the participants by gender and we will divide participants into ‘sitters’ and ‘walkers’ by evaluation of interaction effects in the mixed model. To evaluate the effect of age, we will calculate the difference in the outcome measures between the placebo and pyridostigmine period for each individual. Subsequently, we will investigate if there is a correlation between this difference and age. We will do the same for disease duration and investigate possible covariation.
We will summarise incidence of AEs by treatment group and in all treatment groups combined in frequency tables.
Ethics, dissemination and safety monitoring
This study is registered in the European registry for clinical studies and trials (2011-004369-34; https://www.clinicaltrialsregister.eu) and in the American registry for clinical studies and trials (NCT02941328; https://clinicaltrials.gov). The investigator obtains written informed consent before study participation from participants and from parents if the participant is <18 years old.
The trial is monitored by an external independent party (Jullius Clinical; Broederplein 41–43, 3703 CD Zeist the Netherlands). Because of the short trial period, consecutive monitor visits are only separated by a few months, therefore, monitoring is intense and extensive. Because of the short study period with short visit intervals, mild potential risks of the study medication and intensive monitoring, an interim analysis or safety surveillance by a data safety monitoring board is not indicated. All participants are insured by the sponsor in case of harm due to trial participation.
The study is conducted according to the principles of the Declaration of Helsinki (latest version WMA General Assembly 2008, Seoul) and in accordance with the Medical Research Involving Human Subjects Act. Directly after study inclusion, we assign a random ID code to the participant, which will be used on all documents to ensure confidentiality.
The results of this study will be shared with the academic and medical community, funding and patient organisations in order to contribute to optimisation of medical care and quality of life for patients with SMA.
Strengths and limitations
At the start of this trial, no treatment to cure or slow down SMA was available. Various treatment strategies had been tested to prolong survival in SMA type 1 and improve motor function and strength in SMA types 1–3, but none of them had shown efficacy.55 56
The discovery of structural and physiological abnormalities of the NMJ resulted in new treatment opportunities to improve motor strength, endurance and consequently quality of life. For this purpose, we decided to conduct this trial, with the well-known and safe drug pyridostigmine. It is important to note that even if pyridostigmine is capable of improving the NMJ function and shows to be effective in improving strength and/or endurance, it will not resolve all symptoms of SMA, but hopefully it will improve daily functioning with minimal side effects.
In the autumn of 2016 efficacy of the antisense oligonucleotide nusinersen, defined as improvement on the HINE and Hammersmith Functional Motor Scales in infants and children with SMA, was reported.21 22 The FDA and EMA have approved treatment of patients with SMA types 1–4. However, evidence for effects in patients with milder disease severity and long-standing disease course is currently still lacking and this may complicate reimbursement decisions in at least some countries. While therapy development, including gene therapy,57 is ongoing and promising, there are currently no alternative therapies available. Thus, there remains a need for low cost, easy-to-administer drugs that improve motor function, fatigability and quality of life of patients with longer disease duration who cannot or do not want to be treated with (repetitive) intrathecal injections of nusinersen. More in general, expanding treatment options for all types of SMA in all ages and life stages is essential and pyridostigmine is a well known, safe and low-cost option, stressing the importance of this trial.
One of the major challenges in SMA research is the development and use of outcome measures that can capture the wide variability between and within SMA types and monitor (small) changes of muscle strength, function or fatigability in this slowly progressive disease. The incorrect use of instruments or measurement of irrelevant parameters could result in unnecessary type II errors in trials. The 6 min walk test has been evaluated as an outcome measure for fatigability in SMA,58 however, there is some conflicting evidence for this test.59 Furthermore, we needed an additional test to measure fatigability in upper limbs in patients with SMA who are not able to walk, preferably based on a similar method as the test for patients who are able to walk. Therefore, we developed various new instruments to capture fatigability and objectify endurance capacity (Bartels et al in progress). An obvious limitation of using these tests is that they have not been validated in a large group. This is currently being done parallel to this study. Nevertheless, these tests allow us to investigate the effect of pyridostigmine on endurance in patients with SMA, a dimension of SMA for which outcome measures are currently largely lacking. Similarly, the r9HPT is not a validated test, but data from our previous study show the r9HPT to detect fatigability in patients with SMA type 2, and there was a good test–retest reliability (Stam et al, submitted data). We use the MFM, which has been extensively validated in patients with SMA, as primary outcome measure for motor function.25 26 Another widely used scale is the Hammersmith Functional Motor Scale Expanded.60 To minimise the burden on patients, we selected one of the two scales. Since the MFM was used in several studies, including an international trial at the moment of trial design and start,61 we decided to incorporate it in our trial as well.
Based on previous neurophysiological and clinical studies,13 58 we expect that fatigability is a feature of all SMA types, including milder forms. We will therefore focus on whole-group results, but we additionally plan to stratify participants based on their ability to walk. However, we offer all eligible patients the opportunity to participate and are dependent on the willingness of patients to enrol in this trial. Therefore, it is difficult to predict the number of patients in each stratum, but based on incidence62 and the exclusion criterion of an MFM score >80% we expect more ‘sitters’ than ‘walkers’.
The cross-over design, we use in this study, allows participants to act as their own control and is an ideal design for rare diseases with a wide range of disease severity including SMA, because the unsystematic variance is drastically reduced allowing systematic variance to be detected in a smaller number of participants. Although we cannot exclude external confounders completely since participants are monitored over a period of 4–5 months, in which external factors can be introduced. To minimise the effect of confounders, we ask participants extensively about possible confounding factors such as changes in work or school schedules, lack of sleep and outside temperature. The cross-over design does require specific attention to possible carry-over effect and medication-specific adjustment of the washout period. In our study, the short half-life of pyridostigmine (3–4 hours when kidney function is normal) results in minimal to no carry-over effect with a washout period of only 1 week. Another strength of this study is the use of different outcome measures to evaluate fatigability, (perceived) fatigue and quality of life from several angles, allowing us to take these in consideration when analysing the effect of pyridostigmine.
In conclusion, we believe that we can properly investigate the effect and efficacy of pyridostigmine in this double-blinded, placebo-controlled, cross-over trial and we expect the results of to confirm that pyridostigmine could be used as an (add-on) therapy to improve the function of NMJ defects in patients with SMA resulting in improved strength and/or endurance and/or fatigability.
Acknowledgments
We are grateful to the patients with SMA who participate in this study and the support of the Dutch patient organisation for neuromuscular diseases (www.spierziekten.nl). We would like to thank Professor Jan Verschuuren at the department of Neurology of the Leiden University Medical Center (LUMC), the Netherlands, for his kind advice on pyridostigmine dosing.
References
Footnotes
Contributors Study concept and design were conducted by MS, RIW, CAW and WLvdP. Critical revision of concept and design and intellectual input in the study protocol was done by MS, RIW, CAW, BB, HSG, JFdG, MAGCS, IC, LHvdB and WLvdP. Collection of data is done by MS, CAW, BB, F-LA, LAMO, HSG and LEH. Technical, administrative and material support was provided by F-LA and BB. Drafting of the manuscript was done by MS and RIW. Critical revision of the manuscript was performed by MS, RIW, CAW, BB, F-LA, LAMO, HSG, LEH, JFdG, MAGCS, IC, LHvdB and WLvdP. Study supervision is conducted by LHvdB and WLvdP.
Funding The study is supported by grants from Spieren voor Spieren, Prinses Beatrix Spierfonds, Rotary Bussum and VriendenLoterij. The investigators have full access to the data and have the right to publish this data separate and apart from any sponsor.
Disclaimer Funders did not have any role in the design of the study, collection, analysis, and/or interpretation of data.
Competing interests BB serves on scientific advisory board for Roche Hoffman-La Roche, Zurich. LHvdB serves on the scientific advisory boards for the Prinses Beatrix Spierfonds, Thierry Latran Foundation, Biogen Idec and Cytokinetics; received an educational grant from Baxter International; serves on the editorial board of Amyotrophic Lateral Sclerosis and the Journal of Neurology, Neurosurgery and Psychiatry and receives research support from the Prinses Beatrix Fonds, Netherlands ALS Foundation, The European Community’s Health Seventh Framework Programme (grant agreement no 259867), The Netherlands Organisation for Health Research and Development (Vici Scheme, JPND (SOPHIA, STRENGTH)). WLvdP serves on the scientific advisory boards of Biogen and Avexis and the LMI070 data monitoring committee of Novartis and receives research support from the Prinses Beatrix Spierfonds, Netherlands ALS Foundation and Stichting Spieren voor Spieren.
Patient consent Not required.
Ethics approval The local and national medical ethical committees, Medical Ethical Committee of the University Medical Center Utrecht and Central Committee on Research Involving Human Subjects, respectively, approved the study protocol (dates: 21/04/2015 and 03/11/2014).
Provenance and peer review Not commissioned; externally peer reviewed.