Article Text

Abstract
Introduction Persons with type 1 diabetes require intensive insulin therapy to achieve glycaemic control, but side effects, including hypoglycaemia and weight gain, may reduce treatment compliance. We hypothesise that add-on treatment of the short-acting glucagon-like peptide-1 receptor agonist, exenatide, to insulin therapy in persons with type 1 diabetes will reduce insulin requirements, glycaemic excursions and body weight and improve glycaemic control without increasing the risk of hypoglycaemia. The present article describes a protocol developed to test this hypothesis.
Methods and analysis One-hundred adult persons with type 1 diabetes for more than 1 year, insufficient glycaemic control (glycated haemoglobin A1c (HbA1c) between 58 and 86 mmol/mol) and body mass index >22.0 kg/m2 will be randomised to either exenatide 10 µg three times per day (at meal times) or placebo as add-on therapy to regular basal–bolus insulin treatment for 26 weeks. Primary endpoint is change in HbA1c between the two groups at end of treatment. Secondary endpoints include change in glycaemic excursions (assessed by continuous glucose monitoring); insulin dose; hypoglycaemic and adverse events; body weight, lean body and fat mass; dietary patterns; quality of life and treatment satisfaction; cardiovascular-disease risk profile; metabolomics; and arginine-tested plasma glucose, glucagon and C-peptide levels.
Ethics and dissemination The study is approved by the Danish Medicines Agency, the Regional Scientific Ethics Committee of the Capital Region of Denmark and the Data Protection Agency. The study will be carried out under the surveillance and guidance of the good clinical practice (GCP) unit at Copenhagen University Hospital Bispebjerg in accordance with the ICH-GCP guidelines and the Helsinki Declaration. Positive, negative as well as inconclusive results will be sought disseminated at scientific meetings and in international peer-reviewed scientific journals.
Trial registration number NCT03017352.
- general diabetes
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
First randomised, double-blinded, placebo-controlled trial to investigate a short-acting glucagon-like peptide-1 receptor agonist as add-on therapy in type 1 diabetes.
This study will include normal to overweight persons with insufficient glycaemic control, who represent a large proportion of persons with type 1 diabetes worldwide.
The study is powered to draw conclusions on glycaemic control (as assessed by glycated haemoglobin), glycaemic variability and time spent in near normoglycaemia.
The study may not be sufficiently powered to draw firm conclusions on all secondary endpoints.
Introduction
Background and rationale
Type 1 diabetes is a global disease affecting millions of people with increasing incidence.1 2 The majority of persons with type 1 diabetes do not achieve glycaemic control, and up to 50% are overweight or obese with a body mass index >25 kg/m2.3–6 Intensive insulin treatment is necessary to ensure glycaemic control that delays the onset and slows the progression of microvascular complications, that is, diabetic retinopathy, neuropathy, nephropathy and macrovascular disease.7–9 Failure to achieve glycaemic control may occur due to side effects of intensive insulin treatment, that is, weight gain and hypoglycaemia.10 11 Both weight gain and hypoglycaemia have been shown to reduce treatment compliance. Severe hypoglycaemia is associated with serious physiological and psychological comorbidity and even death.11 Milder hypoglycaemic episodes lead to fear of future episodes, and unwanted weight gain leads to reduced insulin doses.12 13 Overweight itself is unwanted among persons with type 1 diabetes and associated with its own problems, for example, hypertension, cancer and increased cardiovascular disease risk.14
To improve treatment of type 1 diabetes, these problems must always be considered and addressed. Add-on therapy of non-insulin drugs developed for type 2 diabetes has recently gained increasing interest within type-1-diabetes research.15 The incretin hormone, glucagon-like peptide-1 (GLP-1), regulates glucose metabolism through GLP-1 receptor-induced pancreatic and extrapancreatic effects, for example, increased glucose-dependent insulin secretion, lowered postprandial glucagon secretion and reduced rate of gastric emptying.16 Furthermore, GLP-1 promotes satiety and thereby facilitates body weight loss. Several GLP-1 receptor agonists (GLP-1RAs) are used successfully in the treatment of type 2 diabetes—including insulin-treated persons with type 2 diabetes.17 Based on their pharmacokinetic profiles, the GLP-1RAs can be divided into short-acting or long-acting compounds with important between-class differences.
In type 2 diabetes, long-acting compounds exert continuous insulinotropic and glucagonostatic effects. Therefore, they have a greater—and sustained—effect on fasting plasma glucose compared with the short-acting GLP-1RAs. Lowering of fasting plasma glucose is pivotal in insulin-treated type 2 diabetes, and long-acting-compound treatment therefore generally translates into better glycaemic control compared with short-acting GLP-1RAs. In contrast, treatment with short-acting GLP-1RAs exerts potent and sustained slowdown of gastric emptying with an effective lowering of postprandial plasma glucose excursions, an effect lost with long-acting GLP-1RAs due to tachyphylaxis.17 Thus, persons with adequately controlled fasting plasma glucose that are in need for postprandial glucose lowering to achieve glycaemic control will most likely benefit more from a short-acting GLP-1RA compared with a long-acting GLP-1RA.18 In contrast to the different, glucose-lowering effects of the different GLP-1RAs, the body weight-reducing effects of GLP-1RAs seem independent of their pharmacokinetic profile.17
GLP-1RAs provide a valuable treatment concept for persons with type 2 diabetes. Their insulin-independent effects, that is, glucose-dependent glucagon suppression (occurring only at plasma glucose concentrations above 4–5 mmol/L), appetite reduction and deceleration of gastric emptying, make them interesting from a type-1-diabetes management perspective. The long-acting GLP-1RA, liraglutide, was previously examined in several randomised, double-blinded, placebo-controlled trials as add-on treatment in persons with type 1 diabetes. These studies indicated substantial reductions in body weight and total exogenous insulin dose and, in general, moderate improvements in glycaemic control, but at the expense of increased incidences of symptomatic hypoglycaemia and hyperglycaemia with ketosis in the two ADJUNCT studies.19–23
Importantly, the effect of short-acting compounds on postprandial glucose excursions may be of particular interest as several studies have shown a strong correlation between postprandial glucose control and glycated haemoglobin (HbA1c) in type 1 diabetes.18 However, no large controlled clinical trial evaluating the short-acting GLP-1RA treatment effect in type 1 diabetes has been reported. Smaller, mainly mechanistic, studies of exenatide, a short-acting GLP-1RA normally administered two times per day, have shown reductions in postprandial glucose excursions and insulin requirements (0.17–1.19 U/kg/day) together with weight loss (2.8–4.5 kg) and improved, or at least unaltered, glycaemic control.24–26 The main mechanisms for these effects seem to involve deceleration of gastric emptying,27–29 and possibly reduced postprandial glucagon secretion.30 31 Importantly, exenatide given two times per day did not decrease the glucagon response during a hypoglycaemic clamp after 4 weeks of treatment,32 indicating that exenatide’s blood glucose-lowering effects do not compromise the main counter-regulatory effects during hypoglycaemia.
Hypothesis
We hypothesise that add-on therapy of exenatide 10 µg three times per day at main meals to basal–bolus insulin therapy in normal to overweight/obese persons with type 1 diabetes with inadequate glycaemic control (HbA1c between 58 and 86 mmol/mol) will reduce insulin requirements, glycaemic excursions and body weight and improve glycaemic control without increasing the risk of hypoglycaemia.
Objectives and endpoints
The overall objective of the present study is to evaluate the safety and efficacy of the short-acting GLP-1RA, exenatide, administered three times per day (before each main meal) as add-on therapy to standard basal–bolus insulin regimen in persons with type 1 diabetes. The primary endpoint is change in HbA1c after 26 weeks of treatment compared with placebo. Secondary endpoints include changes in glycaemic excursions; insulin dose; hypoglycaemic and adverse events; body weight, lean body mass, fat mass; dietary patterns; quality of life and treatment satisfaction; cardiovascular-disease risk profile; metabolomics; and arginine-tested plasma glucose, glucagon and C-peptide levels (box 1).
Primary and secondary endpoints
Primary endpoint
HbA1c
Secondary endpoints
CGM: Glycaemic variability and time spent in hypoglycaemia, near normoglycaemia and hyperglycaemia
Insulin dose
Hypoglycaemic events
Body weight
BMI
Body composition (hip:waist ratio)
DXA scan: Lean body mass and fat mass composition
Fasting plasma glucose
Dietary patterns
Arginine test: Pre/poststimulatory levels of glucagon, C-peptide and glucose
Cardiovascular disease risk profile: Cholesterol levels, biomarkers, blood pressure and heart rate
Quality of life and treatment satisfaction
Adverse events
BMI, body mass index; CGM, continuous glucose monitoring; DXA, dual-energy X-ray absorptiometry; HbA1c, glycated haemoglobin.
Trial design
The MAG1C trial (Meal-time Administration of Exenatide for Glycaemic Control in Type 1 Diabetes Cases: a randomised, placebo-controlled trial) is a 26 weekinvestigator-initiated, two-armed, parallel group, randomised, double-blinded and placebo-controlled study.
Methods and analysis
In total, 100 persons with type 1 diabetes on basal-bolus insulin therapy will be randomised in a 1:1 ratio to either meal-time exenatide 10 µg three times per day or placebo as add-on therapy to regular insulin treatment. A study-independent person will use a computer-generated randomisation list for treatment allocation. Data will be stored in paper-based case report files (CRF). Double data entry into a digital database with range checks for data values will be used. In case of emergency, unblinding will be made on an individual basis not affecting other study participants. All data will be pseudoanonymised.
Study population
Study participants will be recruited from outpatient clinics in the Capital Region of Denmark. All recruited participants meeting the eligibility criteria at screening will be enrolled in the study and treated for the following 26 weeks at the Steno Diabetes Center Copenhagen, Gentofte, Denmark (box 2).
Eligibility criteria
Inclusion criteria
Type 1 diabetes according to WHO criteria with duration of ≥1 year
Age ≥18 years
BMI >22.0 kg/m2
HbA1c >7.5% and <10.0% at visit 0 (screening)
Able to count carbohydrates
Able to understand the written patient information and to give informed consent
Exclusion criteria
Insulin pump treatment
Hypoglycaemia unawareness (inability to register low blood glucose)
Diabetic gastroparesis
Compromised kidney function (eGFR <60 mL/min/1.73 m2, dialysis or kidney transplantation)
Liver disease with elevated plasma alanine aminotransferase >three times the upper limit of normal (measured at visit 0 with the possibility of one repeat analysis within a week, and the last measured value as being conclusive)
History of acute and/or chronic pancreatitis
Subjects with personal or family history of medullary carcinoma or MEN syndrome
Inflammatory bowel disease
Cancer, unless in complete remission for >5 years
Proliferative retinopathy
Other concomitant disease or treatment that according to the investigator’s assessment makes the patient unsuitable for study participation
Alcohol/drug abuse
Fertile women not using chemical (tablet/pill, depot injection of progesterone, subdermal gestagen implantation, hormonal vaginal ring or transdermal hormonal patch) or mechanical (spirals) contraceptives
Pregnant or nursing women
Known or suspected hypersensitivity to trial product or related products
Receipt of an investigational drug within 30 days prior to visit 0
Simultaneous participation in any other clinical intervention trial
Withdrawal criteria
In case of pregnancy (or desire for pregnancy), female subjects are withdrawn
Lack of compliance to any of the important study procedures in the discretion of the investigator
Onset of any disorder considered to compromise the safety by participating in the study
Unacceptable adverse effects in the discretion of the investigator
Withdrawal on participants request will be accepted at any time without further justification
BMI, body mass index; eGFR, estimated glomerular filtration rate; HbA1c, glycated haemoglobin; MEN, multiple endocrine neoplasia types 1 and 2.
Trial visits and examinations
Study participants will be provided with written and oral information by the investigator prior to obtaining written informed consent at Steno Diabetes Center Copenhagen. At screening (visit 0), information on demography, medical history, smoking/drinking status and concomitant medication will be obtained. Further, a physical assessment will be made including heart rate, blood pressure, body weight, hip:waist ratio and electrocardiography together with blood samples and urine tests (box 3). Six-day continuous glucose monitoring (CGM), together with a 3-day diet recording, will be made before randomisation (visit 1), at week 4 (visit 2) and at end of treatment (visit 4). Participants not familiar with carbohydrate counting will be offered a standard course before entering the study. Blood samples and urine tests will be taken during the trial (box 3). An arginine test and a dual-energy X-ray absorptiometry (DXA) scan will be made at randomisation (visit 1) and at end of treatment (visit 4) (box 3). Insulin doses will be adjusted by the investigator or qualified study personnel during the trial at study visits based on seven-point plasma glucose profiles, CGM and HbA1c. Blood glucose treatment targets will be based on international guidelines,9 that is, preprandial values of 4–7 mmol/L and postprandial values <10 mmol/L. Following randomisation, changes in insulin types are not allowed. The study participants will be asked to fill out questionnaires on quality of life (The Audit of Diabetes-Dependent Quality of Life) and Diabetes Treatment Satisfactory Questionnaire: status (DTSQs) and change version (DTSQc).33 34 Information on adverse events, current medication, basal–bolus insulin dose, hypoglycaemic events and consultation blood pressure and heart rate will be recorded at all visits. Body weight and hip:waist ratio will be measured as well, except on visit 3. Between-visit telephone contacts will be made to ensure the study participants’ safety and compliance together with evaluation of insulin treatment. Further, the study participants will be instructed to contact the study team if any insulin-dosing or glucose control problems occur. All contacts will be recorded in the CRF (figure 1 and table 1). To further attenuate the risk of hypoglycaemia, no insulin is taken on the visit 1 study day and plasma glucose is measured before administration of the first dose of investigational product and 30 min following ingestion of a standardised meal. Next, telephone contacts are made 1 and 2 weeks after randomisation with careful instruction on reporting any hypoglycaemic and hyperglycaemic events. Finally, the investigational product is started at 5 µg and escalated to 10 µg following telephone contact 2, if tolerated. All contacts will be recorded in the CRF (figure 1 and table 1). Finally to ensure compliance, used investigational product cartridges will be collected at study visit.

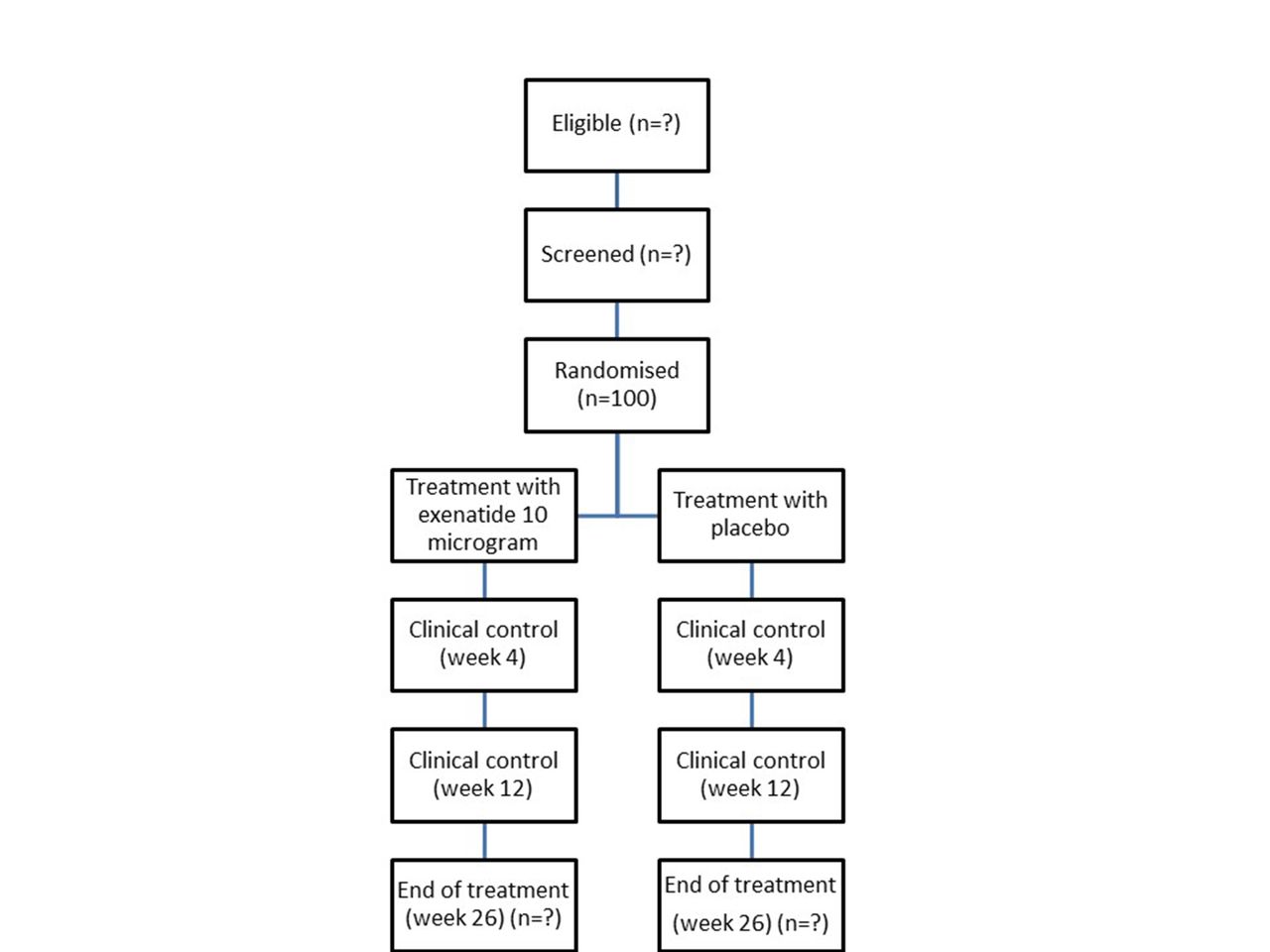{kind=link}
Flowchart.
Blood samples
Screening and control visits
Blood haemoglobin, leucocytes, thrombocytes, plasma glucose, potassium, sodium, creatinine, TSH, cholesterol, triglycerides, ALT, AST, amylase, lipase, serum albumin, total serum-ketones, beta-hydroxybutyrate and acetoacetate
Biobank
CVD markers: HsCRP, pro-BNP
Bone markers: CTX, P1NP, sclerostin, osteocalcin
Inflammation markers: IL-2, IL-6, TNF-α
Urine albumin:creatinine ratio, hCG
Arginine test
Glucagon, C-peptide, plasma glucose
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CTX, C-terminal telopeptide of type 1; CVD, cardiovascular disease; hCG, human choriongonadotropin; hsCRP, high-sensitivity C reactive protein; IL-2, interleukin-2; IL-6, interleukin-6, P1NP, serum type 1 procollagen N-terminal; pro-BNP, prohormone brain natriuretic peptide; TNF-α, tumour necrosis factor-alpha; TSH, thyroid-stimulating hormone.
Trial outline
A substudy on the role of the microbial gut flora, approved by the Danish regulatory authorities and voluntary to participate in, involving the collection of faecal specimens at each study visit will also be conducted.
Patient and public involvement
The MAG1C study aims to attenuate intensive insulin treatment side effects: hypoglycaemia and weight gain. Further, our study drug regimen will, hopefully, make it easier to control blood glucose excursions on a daily basis. We evaluate study participant treatment satisfaction and quality of life through questionnaires during the study period to make sure our results benefit persons with type 1 diabetes. Study participants will be informed of our results in layman’s terms as well as their individual exenatide/placebo assignment by letters following publication. Finally, during the study protocol write-up, a colleague of ours with type 1 diabetes read and commented on the final protocol draft.
Intervention
Name: Byetta (exenatide) or matching placebo.
Pharmaceutical form: exenatide 0.25 mg/mL, 3 mL cartridges in a reusable Ypsopen, for subcutaneous injection. Placebo, 3 mL cartridges in a reusable Ypsopen, for subcutaneous injection.
Pharmaceutical dosage: To minimise the side effect risk, exenatide dose, or placebo, will be increased from initial 5 µg three times per day to full dosage, 10 µg three times per day, 2 weeks after randomisation. The injection must occur within 1 hour before the main meals. Dose increments can be titrated based on the individual study participant’s study drug tolerance, to a minimum of 5 µg three times per day 3 months after randomisation. If not possible at this time, the participant will be withdrawn from the study.
Side effects: Common side effects (1%–10%) include nausea, vomiting, diarrhoea, hypoglycaemia and headache. Study participants will be carefully instructed to avoid dehydration if gastrointestinal side effects occur.
Shipping and packing: All study medication will be produced, blinded, packed and delivered by AstraZeneca, the producer of Byetta.
Sample size
To be able to detect a difference in change in HbA1c (primary outcome) between study arms of 6 mmol/mol with 80% power, a 5% significance level and a presumed 9 mmol/mol SD, 42 persons should be included in each study arm (two-sided test). To allow for a 20% dropout rate, 100 persons in total will be included in the study: 50 in each study arm. The sample size calculation is based on data from a similar study on the GLP-1RA, liraglutide.21 Withdrawn study participants will not be replaced.
Data analysis
The per-protocol study population includes all participants who complete the study with a documented, valid baseline and end-of-treatment assessment of the primary endpoint without any major protocol violations. In case of dropout, last observation is carried forward. The intention-to-treat population includes all randomised persons. Primary-endpoint analysis will be based on the per-protocol population. Absolute differences and adjusted mean changes between groups, together with 95% CI, will be reported. The efficacy analysis will be carried out with a linear mixed-effect model with visit, treatment and their interaction as fixed factors and a random intercept on the person level. Variables that are normally distributed will be presented as mean±SD or SE of the mean. In case of non-normal distribution, non-parametric statistics and log transformation will be used. A two-tailed p value ≤0.05 will be considered statistically significant. Additional analyses will be made from the intention-to-treat population to assess the validity of the per-protocol population conclusions if loss of follow-up occurs. These calculations will include duration in study and reason for discontinuation.
Following completion of last patient last visit, unblinding will be made in two steps. During data analysis, unblinding will be made on group level, that is, participants are assigned to groups 1 and 2. After the prespecified data analysis is completed, the specific treatment group will be revealed.
Ethics and dissemination
We expect the present study to generate important information about the use of short-acting GLP-1RAs as add-on therapy to insulin in persons with type 1 diabetes. We expect to be able to answer two questions relevant for numerous persons worldwide: Will meal-time exenatide 10 µg added three times per day (at each main meal) to regular insulin therapy (1) improve postprandial glycaemic excursions and (2) provide improved, long-term glycaemic control measured as HbA1c and glycaemic variability?
During the study, a physician will follow each participant with careful evaluation of insulin treatment with glycaemic optimisation and study drug safety and efficacy. This is expected to lower the adverse event risk. Exenatide is approved for the treatment of type 2 diabetes by the European Medicines Agency and by the US Food and Drug Administration. Prior studies have shown limited side effects such as nausea, vomiting, hypoglycaemia and headache. Nausea and vomiting, generally transient, usually occur within 3 weeks after treatment initiation. They can be minimised by gradual dose titration, as planned in this study. The hypoglycaemia risk is reduced by insulin dose reduction at study start and by instructing participants in careful blood glucose monitoring. Few cases of acute pancreatitis have been reported in persons with type 2 diabetes using exenatide, but the incidence was similar to the type 2 diabetes background population. Overall, the risk of side effects in this study is expected to be modest. Arginine injection is a well-validated, safe method to evaluate pancreatic alpha and beta cell function, but potentially associated with transient mild flushing, nausea and metallic taste. Vein puncture may cause a short pain, risk of a small haematoma and a minimal risk of puncture site infection. In total, 400 mL blood and 80 mL of urine per person will be collected throughout the study. At the two DXA scans, participants will be exposed to weak X-ray radiation (less than 1 mSv in total). For comparison, the background radiation in Denmark is about 3 mSv per year. The risk of complications, or adverse events, is negligible for all other planned study procedures.
Data will be processed and merged into one or more scientific articles and published in accordance with the CONSORT 2010 statement in international, peer-reviewed scientific journals and presented at national and international scientific meetings. Positive, negative and inconclusive results together with statistical method will be published as soon as scientifically justifiable. AstraZeneca commented on the study design but will have no influence on trial conduction, data analysis, interpretation or publication. All data are owned by the authors, who all have full data access.
Study approval
The MAG1C trial is approved with a current, approved (15 February 2018) study protocol V.1.6. It is registered at ClinicalTrials.gov (NCT03017352). The study will be conducted under the surveillance and guidance of the good clinical practice (GCP) unit at Copenhagen University Hospital Bispebjerg in accordance with the ICH-GCP guidelines and the Helsinki Declaration. The study commenced in January 2017 and is expected to be reported in 2019. Presently (May 2018), 58 study participants have been included in the MAG1C study.
Acknowledgments
We would like to thank Andreas Brønden, MD PhD, as our patient adviser for reading and commenting on the final draft of the study protocol.
References
Footnotes
Contributors NJJ, TFD, AL, TV, HUA and FKK conceived and designed the study. FKK is guarantor of the study and sponsors the trial. NJJ drafted the manuscript and all authors have contributed to the revision of the manuscript and read and approved the final version of the manuscript.
Funding This investigator-initiated research was conducted with support from AstraZeneca.
Competing interests NJJ and TV have no competing interests. TFD has received research support from Novo Nordisk and AstraZeneca, and has received lecture fees from Novo Nordisk. AL has received lecture fees from Novo Nordisk, Boehringer Ingelheim and Eli Lilly. HUA owns stocks in Novo Nordisk and serves in advisory boards for Novo Nordisk and Astra Zeneca. FKK has served on scientific advisory panels and/or speaker’s bureaus for, served as a consultant to and/or received research support from Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Gubra, MSD/Merck, Novo Nordisk, Sanofi and Zealand Pharma.
Patient consent Not required.
Ethics approval Danish Medicines Authority (Eudract-nr.: 2016-001365-92); Regional Scientific-Ethics Committee of the Capital Region of Denmark (H-16034515); Data Protection Agency (2012-58-0004)
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data will be processed and merged into one or more scientific articles and published in accordance with the CONSORT 2010 statement in international, peer-reviewed scientific journals and presented at national and international scientific meetings. Positive, negative and inconclusive results together with statistical method will be published as soon as scientifically justifiable. AstraZeneca commented on the study design but will have no influence on trial conduction, data analysis, interpretation or publication. All data are owned by the authors, who all have full data access.