Article Text

Abstract
Introduction The antiplatelet therapy in the primary prevention of cardiovascular disease in patients with chronic obstructive pulmonary disease (APPLE COPD-ICON2) trial is a prospective 2×2 factorial, double-blinded proof-of-concept randomised controlled trial targeting patients with chronic obstructive pulmonary disease (COPD) at high risk of cardiovascular disease. The primary goal of this trial is to investigate if treatment with antiplatelet therapy will produce the required response in platelet function measured using the Multiplate test in patients with COPD.
Methods and analysis Patients with COPD are screened for eligibility using inclusion and exclusion criteria. Eligible patients are randomised and allocated into one of four groups to receive aspirin plus placebo, ticagrelor plus placebo, aspirin plus ticagrelor or placebo only. Markers of systemic inflammation, platelet reactivity, arterial stiffness, carotid intima-media thickness (CIMT), lung function and quality of life questionnaires are assessed. The primary outcome consists of inhibition (binary response) of aspirin and ADP-induced platelet function at 6 months. Secondary outcomes include changes in inflammatory markers, CIMT, non-invasive measures of vascular stiffness, quality of life using questionnaires (EuroQol–five dimensions–five levels of perceived problems (EQ5D-5L), St. George’s COPD questionnaire) and to record occurrence of repeat hospitalisation, angina, myocardial infarction or death from baseline to 6 months. Safety outcomes will be rates of major and minor bleeding, forced expiratory volume in 1 s, forced vital capacity and Medical Research Council dyspnoea scale.
Ethics and dissemination The study was approved by the North East-Tyne and Wear South Research Ethics Committee (15/NE/0155). Findings of the study will be presented in scientific sessions and published in peer-reviewed journals.
Trial registration number ISRCTN43245574; Pre-results.
- coronary heart disease
- chronic airways disease
- cardiology
- ischaemic heart disease
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Our study will consist of patients who have not been previously targeted in clinical cardiovascular research.
We therefore hope to gather as much information as possible regarding the cardiovascular status of these high-risk patients with chronic obstructive pulmonary disease (COPD) in this proof-of-concept trial.
Our study consists of patients with COPD who have not previously been diagnosed with coronary artery disease (CAD) and yet are at higher risk of CAD, myocardial infarction and excess mortality.
The findings of this study will provide us with detailed information regarding this patient cohort that will enable us to very carefully plan a future large-scale multicentre primary prevention study using antiplatelet therapy in patients with COPD at risk of future cardiovascular events.
This trial is not powered to evaluate hard end points such as death or myocardial infarction.
Introduction
Chronic obstructive pulmonary disease (COPD) is a significant burden on the National Health Service. It is estimated to affect 3 million people within the UK, with approximately 2 million patients remaining undiagnosed.1 We previously examined the evidence for increased cardiovascular (CV) disease risk in patients with COPD. We have reported possible mechanisms linking these two chronic conditions, discussed possible predictors or markers of poor outcomes among patients diagnosed with both COPD and coronary artery disease (CAD), and the therapeutic options aimed at reducing CV risks associated with COPD.2
The prevalence of CAD in patients with COPD has been estimated at 10%–38%,3–8 with 20%–50% of mortality in COPD being related to CV causes.9 10 A retrospective database analysis of pooled data from 2007 to 2010 in the USA has revealed a higher prevalence of cerebrovascular and CV disease in patients with COPD compared with subjects with no COPD, with a short-term risk of 35.8% in subjects with COPD compared with 18.9% in control subjects.11 In the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study, Williams et al demonstrated that coronary artery calcium (CAC) score evaluated on CT scan was higher in patients with COPD than smokers or non-smokers. The presence of CAC is associated with increased dyspnoea, reduced exercise capacity and increased mortality reiterating the fact that the presence of CAD in patients with COPD is associated with poor clinical outcomes.12
Beyond the well-known shared risk factors between COPD and CAD (smoking, low socioeconomic status and sedentary lifestyle), several other mechanisms have been thought to contribute to this increased CV risk. Among these are increased platelet activation and aggregation in patients with COPD, as activated platelets play a key role in the pathogenesis of atherothrombosis. The formation of monocyte-platelet aggregates is a sensitive marker of platelet activation and it takes place in the early stages of the process of development of atherothrombosis.13 Patients with COPD have been shown to have an increased level of platelet-monocytes aggregates compared with control subjects, with further increase in the level of these aggregates during an acute exacerbation.14
Recently, the Study to Understand Mortality and Morbidity (SUMMIT) trial evaluated whether inhaled treatment with a combined treatment of the corticosteroid, fluticasone furoate and the long-acting β-agonist, vilanterol could improve survival compared with placebo in patients with moderate COPD and heightened CV risk. In this large trial of 16 485 participants, treatment with fluticasone furoate and vilanterol did not affect mortality or CV outcomes. This suggests that treatments targeting inflammation in the lung and attempting to improve lung function do not convey any beneficial effects on overall mortality and CV outcomes. Thus, ongoing clinical research is required to evaluate strategies to improve CV outcomes in this high-risk patients with COPD in whom the CVD burden is very high.15–17
Antiplatelet therapy (APT) plays a crucial part in the management of acute coronary syndrome (ACS).18 Recent reports have shown that inhibition of the purinergic receptor P2Y12 is associated with a marked reduction in platelet reactivity, and has anti-inflammatory effects.19 The effects of P2Y12 receptor antagonists beyond platelet inhibition have been previously reviewed in detail.20 In an analysis from the Platelet Inhibition and Patient Outcomes (PLATO) trial, fewer on-treatment pulmonary adverse events (AEs) occurred in the ticagrelor group compared with the clopidogrel group, with fewer deaths following these AEs, particularly in those who remained on study medication 3 days after AE onset.21 Prior data suggest that ticagrelor also influences host defence against bacterial lung infection through adenosine-mediated neutrophil chemotaxis.22 Furthermore, recent studies demonstrated the safe use of ticagrelor in patients with mild-to-moderate COPD with very low rates of bronchospasm.23 There was a similar efficacy benefit and safety profile of ticagrelor compared with clopidogrel with low rates of discontinuation of ticagrelor due to dyspnoea.24 Importantly, blocking ADP receptors with antagonists protects against cigarette smoke-induced lung inflammation and development of emphysema in mice suggesting a potential additional benefit of ADP blockade. A previous observational study showed that the use of aspirin or clopidogrel in patients with acute exacerbations of COPD correlated with a reduction in 1-year mortality (OR 0.63; 95% CI 0.47 to 0.85, p=0.003).25
These additional effects of P2Y12 inhibitors offer the potential for primary prevention in a population at high risk for CV disease such as patients with COPD by reducing inflammation with resultant reduction in CV risk. However, our unpublished database review of 24 000 patients with COPD show very few patients with COPD are receiving APT treatment despite the association of increased platelet activity, inflammation and increased CV events in patients with COPD. Although APT using aspirin and ticagrelor (a novel potent antiplatelet agent P2Y12 receptor antagonist) is now used in the management of patients with ACS, its role in reducing AEs among patients with COPD at risk of cardiac events without ACS has not been previously evaluated in randomised controlled trials. Therefore, we undertook this proof-of-concept trial to evaluate the effect of APT (aspirin, ticagrelor) on platelet function and explored its effect on the inflammatory markers in patients with COPD with no history of CAD.
Hypothesis
Treatment with antiplatelet drugs will result in significant platelet inhibition, improvement in inflammatory markers of CV disease burden and lung function in patients with COPD.
Primary objective
To investigate if treatment with APT will produce the required response in platelet function measured using the Multiplate test in patients with COPD.
Secondary objectives
The secondary objectives of the APPLE COPD trial consists of the following:
To investigate if treatment with APT will lead to reduction in inflammatory marker levels including fibrinogen, high-sensitivity C reactive protein (hsCRP), tumour necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), myeloperoxidase (MPO).
To investigate if treatment with APT will lead to improvement in spirometry (forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC)), vascular stiffness and carotid intima-media thickness (CIMT).
To investigate if treatment with APT will lead to improvement in quality of life.
To record AEs including breathlessness, occurrence of repeat hospitalisation, angina, myocardial infarction or death from baseline to 6 months following treatment with aspirin and/or ticagrelor in patients with COPD.
Methods
The APPLE-COPD trial is a 2×2 factorial, randomised, controlled, participant, investigators and assessors-blinded proof-of-concept trial, as illustrated in figure 1.

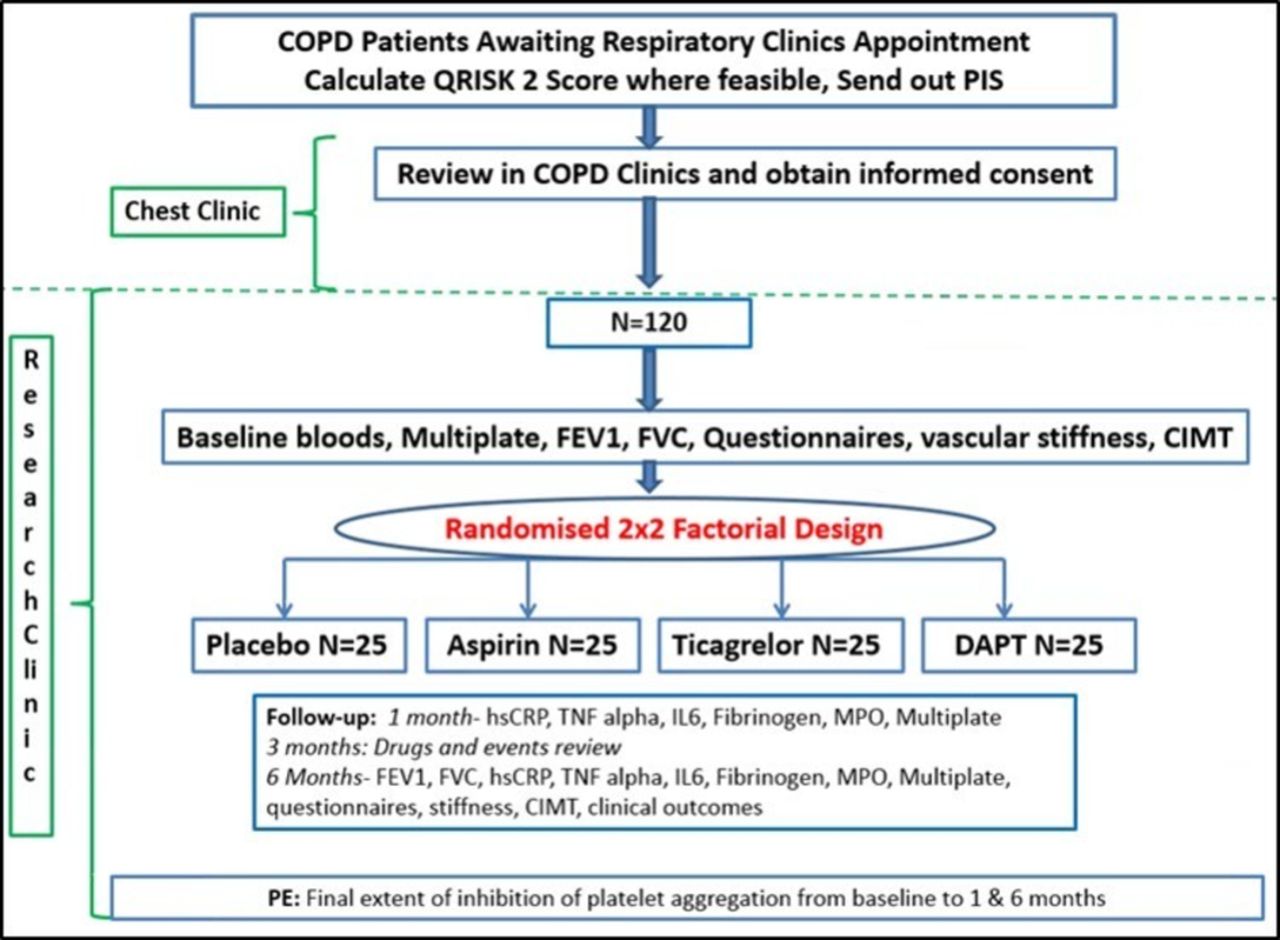{kind=link}
APPLE COPD study flow chart. CIMT, carotid intima-media thickness; FEV1, forced expiratory volume in 1s; FVC, forced vital capacity; hsCRP, high-sensitivity C reactive protein; IL-6, interleukin-6; MPO, myeloperoxidase; PIC, patients’ identification centre; TNF, tumour necrosis factor.
Identification of participants
Patients from respiratory outpatient clinics in the Royal Victoria Infirmary, Freeman Hospital and patient identification centres (PIC) in primary care in the area of Newcastle upon Tyne, UK are included in this trial. In addition, Queen Elizabeth Hospital in Gateshead, Darlington Memorial Hospital and University Hospital of North Durham, UK are also used as PIC sites. The start date for this trial is August 2015 and the original funder proposed end date is December 2018. In the above centres, patients with COPD are screened to determine their eligibility. Patients are then approached by a member of the research team who provides the participant with study information and a patient information sheet (PIS). Patients' details (eligibility, reason for ineligibility, contact details as well as appointment dates and times) are entered on a password protected screening log. Patients are contacted by a member of the research team and those who agree to participate in the study are given an appointment to attend for their baseline visit and subsequent visits at 1 month, 3 months and 6 months in the National Institute of Health Research Clinical Research Facility in the Royal Victoria Infirmary, Newcastle upon Tyne, UK. The inclusion and exclusion criteria are displayed in box 1.
Inclusion and exclusion criteria
Inclusion criteria:
Abnormal spirometry with forced expiratory volume in 1 s (FEV1)<80% and FEV1/forced vital capacity ratio <70% of predicted.
Smoking history that is 10 pack-years or greater (current or ex-smokers can be included).
Have capacity to consent.
Exclusion criteria:
Age<18 years.
Any condition that is being concurrently treated through anticoagulation or antiplatelet therapy (APT) including aspirin (any form of aspirin) or ticagrelor (atrial fibrillation, deep vein thrombosis, valve prosthesis, recent myocardial infarction, use of drug-eluting stents).
Other specific contraindications to management with antiplatelet medication (bleeding risks, allergies).
Any contraindication for aspirin and ticagrelor use.
Other concurrent terminal illnesses with life expectancy <1 year (congestive cardiac failure, carcinoma, etc).
Current involvement in another clinical trial or exposure to another investigational medicinal product within the previous 30 days.
Chronic obstructive pulmonary disease with an atypical cause (eg, A1-antitrypsin deficiency).
Patients who are unable to provide informed consent.
Planned/expected major surgery within next 9 months where APT would be ceased.
Pregnancy, planned pregnancy or current breast feeding.
Intervention
Aspirin is administered as a 75 mg once-daily dose and ticagrelor administered as a 90 mg twice-daily dose for 6 months. The bases for the investigational medicinal products (IMPs) are Brilique (ticagrelor) 90 mg (composition: ticagrelor, mannitol, dibasic calcium phosphate, sodium starch glycolate, hydroxypropyl cellulose and magnesium stearate) and placebo tablets (lactose monohydrate, microcrystalline cellulose and magnesium stearate) supplied by AstraZeneca as well as commercially available aspirin 75 mg tablets (aspirin, maize starch, E553). MODEPHARMA (London, UK) is responsible for arranging the aspirin placebo manufacturing and labelling/randomised packaging of all IMPs and final qualified person (QP) release for clinical trial use. Active and placebo study medication are provided as a 3-month supply. Study medication is prescribed by the principal investigator or the named study clinical research associate according to the protocol, and dispensed to the patient according to local pharmacy policy. Patients in possession of their study medication shall return all trial supplies in their original packaging (even if empty) to the research team at 3-month and 6-month follow-up visits.
Study drug must be discontinued if, participant develops thrombolysis in myocardial infarction (TIMI) major bleed, decides they no longer wish to continue or cessation of study drug is recommended by the investigator. The investigator also has the right to withdraw patients from the study drug in the event of intercurrent illness, AEs, serious AEs, suspected unexpected serious adverse reactions, protocol violations, administrative reasons or other reasons.
All patients will be reviewed by the research team at 1 month, 3 months and at 6 months to ensure and enhance the likelihood of good compliance. Compliance with study medication will be assessed by checking and recording the remaining number of capsules after each visit/at the final visit. Study drug accountability will be assessed and documented by local pharmacy. The clinical team will also perform a quick review of any returned study medication at each study visit to identify any obvious compliance concerns and address these immediately with the participant.
Participants will continue to take medications for other conditions as normal, and a complete listing of all concomitant medication received during the treatment phase are recorded in the electronic case report forms (eCRF).
Primary outcome
The primary outcome for this study consists of inhibition (binary response) ofASPI and ADP-induced platelet function at 6 months. ADP response is defined as ADP <46 and ASPI response is defined as ASPI <40. For patients randomised to ticagrelor, response is according to ADP response and for patients randomised to aspirin, response is evaluated according to the ASPI response.
Secondary outcomes
We will evaluate the following secondary outcomes measures: changes in inflammatory markers including fibrinogen, hsCRP, TNF-α, IL-6, MPO from baseline to 6 months; changes in CIMT and non-invasive measures of vascular stiffness from baseline to 6 months; and changes in quality of life using questionnaires (EQ5D-5L, St. George’s Respiratory Questionnaire for COPD (SGRQ-C)) at baseline and at 6 months. In addition, although this study is not powered to measure hard end points, at the time of the start of this study there were not adequate information available in the literature on the CV event rate. Therefore, in addition in our study we recorded the occurrence of repeat hospitalisation, angina, myocardial infarction or death at 6 months. However, we have noted the incidence of CV events described in the more recently published SUMMIT trial, which evaluated the effect of inhaled corticosteroid and long-acting beta-agonist on survival in patients with moderate COPD and heightened CV risk.15
Safety outcomes
The downside of dual APT is the occurrence of bleeding, which in itself can lead to excess mortality.26 27 Therefore, we monitor and evaluate the bleeding events very carefully in our trial. The primary safety outcome will be rates of major and minor bleeding as defined by the TIMI scale28 and the Bleeding Academic Research Consortium (BARC) definition.29 In addition, given ticagrelor can lead to increase in dyspnoea due its adenosine-mediated effects, we also monitor decline in FEV1, FVC and Medical Research Council (MRC) dyspnoea scale (breathlessness) as a safety outcome measure.
Baseline data collection
At the baseline research clinic, the patients undergo the following procedures by the research team: QRISK2 score using an online calculator (http://www.qrisk.org/); 30 mL blood sample to measure platelet reactivity and biomarker levels; lung function tests (FEV1 and FVC) using spirometry; ultrasound scan to measure CIMT; vascular stiffness using the Vicorder device (Skidmore Medical, Bristol, UK) and quality of life (EQ5D-5L and SGRQ-C) and MRC dyspnoea scale. All data are collected in eCRF using MACRO (Elsevier, Amsterdam, The Netherlands). No patient identifiable data are entered and user access is password protected.
Platelet function testing
High on-treatment platelet reactivity despite use of P2Y12 antagonists is associated with adverse cardiac events.30 A previous study showed that patients with COPD who underwent percutaneous coronary intervention have higher ‘on-treatment’ platelet reactivity compared with patients with no COPD and suggested that this could contribute to the high CV mortality in these patients.31 Aspirin inhibits platelet-mediated atherothromboembolic events by irreversible inhibition of platelet cyclooxygenase-1 with subsequent prevention of the synthesis of the potent pro-aggregatory prostanoid thromboxane A2. Despite aspirin therapy, a large number of patients continue to experience atherothromboembolic events, so-called ’aspirin treatment failure', and this is multifactorial in aetiology. Approximately 10% do not respond appropriately to aspirin in a phenomenon known as ’aspirin resistance'.32 The beneficial effect of aspirin for the primary prevention of CVD is modest and occurs at doses of 100 mg or less per day. Older adults seem to achieve a greater relative myocardial infarction benefit.33 In the APPLE COPD trial, we evaluate the effect of APT in patients with COPD on platelet function. Importantly, this trial will allow us to evaluate compliance of therapy in this patient cohort. Of note, in the SUMMIT trial 23%–29% of patients withdrew investigational product.15
Multiple electrode aggregometry enables rapid assessment of platelet function using a whole blood sample. The signal reaction in multiplate analyser is triggered by the adhesion of activated platelets to the surfaces of the sensor electrodes, which induces an increase of electrical resistance. Each test cell incorporates two pairs of sensors serving as a built-in quality control.34 Multiplate analyser, consumables and reagents have been provided by Roche Diagnostics (Roche Diagnostics, West Sussex, London, UK). The multiplate analyser has an electronic pipette and by using a predefined pipette programme, 300 µL of whole blood from a citrate sample is diluted with warm normal saline (0.9%). For each sample, three assays of platelets reactivity are carried out: (1) ASPI test using arachidonic acid to determine the level of inhibition of cyclooxygenase-induced platelet function in accordance with the proposed aspirin effect; (2) ADP test to determine inhibition of ADP-induced platelets aggregation, which is in accordance with the hypothesised ticagrelor effect; (3) the Thrombin receptor-activating peptide (TRAP) test to determine platelet reactivity via stimulation of the thrombin receptors is used to ensure the reliability of the results given by the analyser, given patients will not be on any glycoprotein IIb/IIIa antagonists to inhibit platelet reactivity. The results are represented by a graphic curve of aggregation (AU)/min. The parameters of the results are: the velocity (AU/min), the aggregation (AU) and area under the curve expressed in AU×min or U (10 AU×min=1U).
Inflammatory biomarker analysis
Neurohormonal activation and systemic inflammation have been suspected to play a significant role in increasing the risk of CV disease.35–37 The exact mechanism by which systemic inflammation develops in patients with COPD is not yet well understood. Some studies have suggested this could be due to the release of cytokines and oxygen radicals from the lungs into the systemic circulation, while others attribute this to the hyperactivity of the sympathetic nervous system leading to the activation of a peripheral inflammatory response.38 Biomarkers of systemic inflammation, including fibrinogen, CRP,36 IL-6, IL-8 and TNF-α,39 40 are higher in patients with stable COPD as compared with control subjects. The release of oxygen radicals may promote the oxidation of low-density lipoproteins and the formation of foam cells increasing the risk of CAD as well as inducing a systemic immune response.41 Given ticagrelor has previously been shown to have anti-inflammatory properties20 and also associated with reduced mortality following pulmonary AEs and sepsis21 and the fact that patients with COPD have heightened inflammatory burden, we wanted to evaluate the effect of ticagrelor on the various inflammatory markers and CV disease burden in this trial.
Blood samples are collected at baseline, 1-month and 6-month visits and tested for full blood count, lipid profile, hsCRP, fibrinogen, TNF-α, MPO, IL-6 and microRNA.
Vascular stiffness
Arterial stiffness is another emerging predictor of the risk of CVD. Arterial pulse wave velocity (aPWV) is a widely used method of assessing arterial stiffness. A recently published meta-analysis of 27 longitudinal studies (>20 000 participants) revealed a strong and positive relationship between aPWV and CV morbidity and mortality.42 The severity of emphysema was found to be independently associated with higher arterial stiffness,43 44 which could possibly be one of the mechanisms of increased risk of CVD in patients with COPD independent of systemic inflammation. Some studies showed aPWV to be higher in patients with COPD but not to be related to the level of inflammatory markers; however, age and blood pressure were found to be determinants of aPWV in these patients.45 46 We evaluated the baseline vascular stiffness in patients and also repeated these measures at 1 month and 6 months. It is unlikely that APT will lead to significant effect on the vascular stiffness. However given the anti-inflammatory properties of ticagrelor, we wish to explore this further in our study.
aPWV is measured using Vicorder device (Skidmore Medical), which relies on pressure sensors. This is a non-invasive and reproducible method that can be performed at the bedside using Vicorder software (Skidmore Medical). Participants will be asked to lie on a bed at approximately 45 degrees angle with blood pressure cuffs placed on the arm, upper leg and neck to measure brachial, femoral and carotid artery pressures, respectively. The distance between the sternal notch to the femoral cuff will be used in assessment of aPWV and from sternal notch to umbilicus to assess brachiofemoral pulse wave analysis (PWA). PWA assessment includes measurement of the pulse pressure (PP), augmentation index, which is the difference between the first and second systolic peaks relative to PP and is used as a surrogate measure of arterial stiffness,47 and the augmentation pressure in addition to ankle brachial pressure index. PP (systolic minus diastolic blood pressure) can also be used as a surrogate measure of arterial stiffness. A PP wave is generated by each contraction of the left ventricle and it precedes blood flow down the aorta. In normal subjects, the PP wave propagates down the aorta to the peripheral arterioles and capillaries. At the level of the capillaries, part of this wave is transmitted down these small blood vessels and is responsible for the continuous low pulsatile blood flow to the tissues; while part of the wave is reflected back to the left ventricle. The stiffer the arteries and mainly the aorta the faster this reflected wave travels back to the left ventricle causing an increase in the afterload and systolic blood pressure and may result in reduction in the diastolic coronary blood flow.48 49
Carotid intima-media thickness
A previous study that compared hsCRP and CIMT in newly diagnosed untreated patients with COPD versus subjects without COPD showed CIMT and hsCRP to be significantly higher in the COPD group (p<0.05).50 A large study which assessed the association between hsCRP, fibrinogen and CIMT in 2502 patients with COPD concluded that these two inflammatory biomarkers are related to CIMT in a multivariate-adjusted analysis and smoking was associated with a higher CIMT in men.35 In the APPLE trial once again given the potential anti-inflammatory effects of ticagrelor, we explored the effect of APT on CIMT in patients with COPD. Long-term follow-ups will be required to show significant impact on CIMT. However, this study was not planned as a long-term follow-up study but a study to understand the basic mechanisms that might help us guide in the planning of future studies.
CIMT is measured using Xario XG ultrasound machine with a vascular probe to obtain ECG gated images of both the right and left common carotid arteries. The visualised CIMT of both near and far walls are measured at approximately 1 cm proximal to the carotid bulb using manual calibration method. Readings of maximum and average CIMT are obtained.
Lung function testing
One of the hallmarks of COPD is an accelerated decline in lung function, as measured by spirometry. Inflammation, oxidative stress and other pathways are hypothesised to be important in this deterioration. We wanted to evaluate if APT therapy impacting the inflammatory burden in the lungs will lead to improvement in lung function measured using spirometry. Spirometry is performed by a trained member of the research team at baseline (unless previously conducted in the 30 days prior to baseline visit during a routine clinic appointment) and at 6-month visit using a Vitalograph Gold Standard Bellows Spirometer.
Quality of life questionnaires
Quality of life is impaired in patients with COPD.51 Improvement in lung function could potentially lead to improvement in quality of life in these patients. Two questionnaires are used to assess the quality of life at baseline, 1 month and 6 months visits. These are the SGRQ-C and the EQ5D-5L. SGRQ-C is the shorter version of the SGRQ, which is a research tool that has been shown to have good discriminative properties and is responsive to therapeutic trials; strong correlation has been demonstrated between SGRQ-C scores and the severity of COPD.51 52 The EQ5D-5L Questionnaire assesses the self-reported health status at the time of completion of the questionnaire.53 In the present study, we will be able to evaluate the changes in quality of life in the different treatment groups.
Follow-up
Compliance with study medications and the occurrence of AEs are continuously assessed throughout the study at 1 month, 3 months and 6 months. In addition, all study-related procedures described above are measured at 1 month and at 6 months. Schedule of events is shown in table 1.
Schedule of events
Sample size calculation
For each research question, a Fleming A’Hern54 early phase design is used to investigate the roles of (i) aspirin and (ii) ticagrelor as being individually worthy of further investigation. To determine whether aspirin or ticagrelor is likely to meet a minimum level of efficacy worthy of further investigation, the Fleming A’Hern54 design requires specification of the response rates at 6 months above and below which would indicate whether to proceed or not proceed to a phase III trial. These rates have been chosen based on a study by Perl et al,55 who report a response rate for ticagrelor and prasugrel at 1 month of 100% and 91.3%, respectively in a non-randomised comparison. The APPLE COPD trial design assumes a 6-month rate would be high, similar to the reported 1-month rate of Perl et al. and as such assumes a response rate to reject either treatment (p0) <65% and a response rate to investigate either treatment further (p1) >80%. The design of the trial focuses on demonstrating that a response rate of 80% is plausible and that the response rate is at least >65%. For each research question, the justification to investigate either treatment further is based on observing a minimum number of responses in each interventional arm, referred to as the critical number. Due to minimal background evidence, the control arms provide a direct and unbiased benchmark. As an early phase trial, the error levels have been inflated beyond those expected for a definitive large phase III study but restricted to an acceptable level for an early phase trial of 12% alpha (type 1) and 12% beta (type 2). With these stated parameters, the target recruitment is calculated as 50 patients per arm for each comparison within the 2×2 design: (i) aspirin (n=50) versus no aspirin (n=50); (ii) ticagrelor (n=50) versus no ticagrelor (n=50), as below. Fifty was the smallest number which satisfies the design criteria above. In this trial, the ‘no aspirin’ and ‘no ticagrelor groups’ provide an unbiased control benchmark response rate due to minimal background evidence. As this trial is investigating outcomes at 6 months following randomisation, the sample size is adjusted for an anticipated 15% dropout rate within the 6-month timeframe, rounded to a target recruitment of 120 patients. The estimated dropout rate will be monitored throughout the recruitment period. The Fleming A’Hern design54 would indicate no further investigation of each treatment individually if the observed number of responses in each intervention arm (for aspirin and ticagrelor separately) is less than the critical number.
Recruitment
Patients with COPD will be identified in PIC. The PIC will provide information about the study by handing out the PIS to eligible patients and/or will advertise the opportunity to participate in the study via posters in their waiting rooms. The patients will be invited to participate in the study by the research team (PIs, clinical research associate or research nurse) after being given adequate information on the study.
Randomisation
Consenting patients who fulfil the inclusion and exclusion criteria are randomised using the Newcastle Clinical Trials Unit secure web-based system. Participants are allocated into one of four groups to receive either aspirin plus placebo, ticagrelor plus placebo, aspirin plus ticagrelor or placebo only, using a stratified blocked treatment allocation (permuted random blocks of variable length). The randomisation list was generated by an independent statistician. Stratification is based on the important confounder of age (dichotomised as ≤65 years, >65 years). Each patient is allocated a unique trial number at the point of randomisation. Treatment allocation is double-blinded and as such the randomisation procedure generates a treatment pack number for each participant that links to the corresponding allocated study drug. This automated procedure will produce a pharmacy prescription including the patient’s unique trial number and treatment pack number to ensure the study pharmacist dispenses the correct trial medication.
Blinding
Assignment to either active or placebo arm will be blinded to both the participant and investigators/assessor (double-blind). The need for emergency unblinding is felt unlikely and will preferably be avoided. In case of emergency (bleeding), it will be assumed that patient is on dual APT. In the event of bleeding on such medications in routine practice the therapies are simply withheld. No specific therapies are instituted in this setting after initiating simple support measures to reduce bleeding including pressure. In the event of any bleeding deemed clinically significant by the healthcare professional seeing a study participant (either as part of the study team or as part of emergency services), the patient will be advised to provide the patient participation card. This will contain both emergency contact numbers for the study team (to be contacted if deemed needed) and also clear instructions on stopping the trial medications.
Data collection and management
The strategy of reviewing all patients at 1, 3 and 6 months will promote participant retention and complete follow-up. Should a patient withdraw from study drug only, efforts will be made to continue to obtain follow-up data, with the permission of the patient. Data will be entered directly onto a data management system by the research team and stored on the Newcastle Clinical Trials Unit’s MACRO database, provided by Elsevier’s Software as a service system MACRO is a secure validated clinical data management system. Data will be handled, computerised and stored in accordance with the Data Protection Act 1998. No participant identifiable data will leave the study site. The quality and retention of study data will be the responsibility of the chief investigator. All study data will be retained in accordance with the latest directive on Good Clinical Practise (GCP) (2005/28/EC) and local policy.
Patient and public involvement
In this trial, patient and public were not involved.
Statistical analysis
Descriptive statistics will be tabulated by comparative group and overall. Continuous measures will be presented as mean (SD) or median (IQR). As a 2×2 factorial design, the primary analyses are based on a comparison of: aspirin versus no aspirin and ticagrelor versus no ticagrelor. For each analysis, the primary outcome measure is platelet response rate measured at 6 months (1 month response rate is assessed as a secondary outcome measure). Response rate will be calculated on the intention-to-treat (ITT) dataset as the total number of patients responding as a proportion of all patients randomised (patients with missing primary outcome data are classed as non-responders) and reported descriptively as both a rate and unadjusted OR with 95% CIs. A sensitivity analysis will be carried out on the modified ITT dataset. Response rate will be calculated on the modified ITT dataset as the total number of patients responding as a proportion of all patients randomised with a primary outcome measurement at 6 months. Any patients who are not assessable at 6 months will be classed as missing and removed. A sensitivity analysis will be carried out on the per protocol (PP) set where the response rate will be calculated on the PP dataset as the total number of patients responding as a proportion of the number of patients within each comparative group of the PP dataset. Any patients with missing primary outcome data, who are protocol deviators are removed. Secondary analyses will be based on adjusted OR of the treatment effect adjusted by the stratification variables (age) estimated in a multivariable logistic regression model and 1 month reported response rate. AEs will be reported descriptively as the number of events, and number of unique patients experiencing an AE as a proportion of the total number of patients starting treatment. Biological outcome measures will be presented graphically and descriptively as medians (with IQRs). Patient quality of life will be scored according to instructions for the individual questionnaires and reported graphically and descriptively as means (with SD). Due to the randomisation process, platelet reactivity is assumed balanced at randomisation. Previous literature is minimal and as such this trial is designed using early phase methodology with the aim to investigate levels of activity worthy of further investigation in later studies. As such, the trial is not powered for any hypothesis testing.
Trial monitoring
Monitoring of study conduct and data collected will be performed by a combination of central review and site monitoring visits to ensure the study is conducted in accordance with GCP. Study site monitoring will be undertaken by Newcastle Clinical Trials Unit following risk-based assessment approved by sponsor. The main areas of focus will include consent, serious AEs, essential documents in study files, drug accountability and management.
The trial may be prematurely discontinued on the basis of new safety information, or for other reasons given by the Trial Oversight Committee and/or Trial Management Group, sponsor, regulatory authority or ethics committee concerned. The Trial Oversight Committee will advise on whether to continue or discontinue the study and make a recommendation to the sponsor. If the study is prematurely discontinued in terms of patient recruitment, participants enrolled to date will be informed, no further participants will be randomised and enrolled participants will be followed up.
Harms
Most AEs and adverse drug reactions that occur in this study, whether they are serious or not, will be expected treatment-related effects due to the drugs used in this study. All non-serious adverse reactions will be recorded at 6 months follow-up, and serious AEs will be recorded throughout the duration of the trial until 4 weeks after trial therapy is stopped. The Medicines and Healthcare products Regulatory Agency (MHRA) and main Research Ethics Committee (REC) will be notified by the sponsor via the electronic MHRA reporting system of all suspected unexpected serious adverse reaction (SUSARs) occurring during the study according to the following timelines; fatal and life-threatening within 7 days of notification and non-life threatening within 15 days. All investigators will be informed of all SUSARs occurring throughout the study on a case-by-case basis.
Auditing
The study is subject to inspection and audit by Newcastle upon Tyne Hospitals NHS Foundation Trust under their remit as sponsor, and other regulatory bodies to ensure adherence to GCP. The investigator(s)/institutions will permit trial-related monitoring, audits, REC review and regulatory inspection(s), providing direct access to source data/documents.
Ethics
The conduct of this study will be in accordance with the recommendations for physicians involved in research on human subjects adopted by the Declaration of Helsinki (1996). Clinical Trial Authorisation from the MHRA and NHS R&D approval (Newcastle upon Tyne Hospitals NHS Foundation Trust) were also obtained prior to commencement of the study. Information sheets (see online supplementary appendix) are provided to all eligible subjects and written informed consent obtained prior to any study procedures.
Supplementary file 1
Protocol amendments
Protocol amendments were agreed on by Newcastle upon Tyne Hospitals NHS Trust and AstraZeneca, and approved by the North East-Tyne and Wear South Research Ethics Committee prior to implementation. In the initial protocol version, we had intended to continue collecting the AEs data up to 1 year. However, in the latest approved protocol amendment version we decided to collect data up to 6 months only.
Consent
One of the research team members will approach the patients to see if they are agreeable to take part in the study. Informed consent discussions will be undertaken by appropriate site staff (as per delegation log), including medical staff and research nurses, with opportunity for participants to ask any questions. Following receipt of information about the study, participants will be given reasonable time to decide whether or not they would like to participate. Those wishing to take part will provide written informed consent by signing and dating the study Patient Informed Consent Form, which will be witnessed and dated by a member of the research team with documented, delegated responsibility to do so.
Confidentiality
All personal data will be regarded as strictly confidential. To preserve anonymity, any data leaving the site will identify participants by their initials and a unique study identification code only. The study will comply with the Data Protection Act, 1998. All study records and Investigator Site Files will be kept at site in a locked filing cabinet with restricted access. All laboratory samples will be labelled with a unique study identification number and patient date of birth only (linked in anonymised form).
Access to data
Chief investigator, co-investigators and the trial team will have access to the data sets, which are password protected.
Ancillary and post-trial care
The Newcastle upon Tyne Hospitals NHS Foundations Trust is sponsor and through the sponsor, National Health Service (NHS) indemnity is provided in respect of potential liability and negligent harm arising from study management. Both sites within this study are hospitals within Newcastle upon Tyne Hospitals NHS Foundations Trust and NHS indemnity is provided in respect of potential liability and negligent harm arising from study conduct. Indemnity in respect of potential liability arising from negligent harm related to study design is provided by NHS schemes for those protocol authors who have their substantive contracts of employment with the NHS and by Newcastle University Insurance schemes for those protocol authors who have their substantive contract of employment with the Newcastle University.
Dissemination
The data will be the property of the chief investigator and co-investigators. Publication will be the responsibility of the chief investigator and published under the authorship agreed with the co-investigators and the trial team who fulfil the ICMJE criteria for authorship. It is planned to publish this study in peer-reviewed journals and to present data at national and international meetings. Results of the study will also be reported to the sponsor and funder. Participants will be informed about their treatment and their contribution to the study at the end of the study, including a lay summary of the results.
Conclusion
This trial is the first study to evaluate APT specifically ticagrelor in patients with COPD with no history of CAD. This study will enable a detailed understanding of the baseline CV disease burden in patients with COPD and the response rate to APT in terms of platelets inhibition. In addition, the effect of APT on inflammatory and CV disease burden is explored. This will help us to very carefully plan future large-scale multicentre primary prevention study among patients with COPD evaluating hard end points such as myocardial infarction and mortality.
Trial management group
Dr Vijay Kunadian (chief investigator); Dr Anthony De Soyza (principal investigator); Professor Deborah Stocken (senior statistician, co-investigator); Professor Andrew Fisher (co-investigator); Dr Nina Wilkinson (trial statistician); Dr Graham Burns (investigator); Ms Nicola Howe (database manager); Sean Scott (sponsor representative); Professor Elaine McColl (co-investigator); Dr Jared Thornton, Dr Lesley Hall (senior trial managers); Dr Alexander von Wilamowitz-Moellendorff, Andrea Bell (trial managers); Dr Eva-Maria Holstein (trial administrator); Laura Robertson (trial secretary).
Trial Oversight Committee
Professor Stephan James (Chair) Uppsala Clinical Research Centre, Uppsala University, Sweden; Dr Neil Swanson, the James Cook University Hospital Middlesbrough UK; Dr Alex McConachie, Institute of Health and Wellbeing, Robertson Centre for Biostatistics, University of Glasgow, UK; Dr John Hurst, Centre for Inflammation and Tissue Repair, University College London.
Trial sponsor
Newcastle upon Tyne Hospitals NHS Foundation Trust. R&D reference 7356.
Trial conduct
Newcastle Clinical Trials Unit Registration number 22.
Supplementary file 2
Acknowledgments
Chief Investigator: VK. Principal Investigator: ADS. The authors are grateful to AF, GB, Dr Sophie West, Dr Stephen Bourke and Dr Jim Lordan for allowing their patients to be included in this trial. Clinical Research Fellows: HA, Dr Sophie Gu, Adam Miller, DC. Primary Care team: Laura Renwick and Sally Dunn. All GP practices in the North East and Cumbria Primary Care Clinical Research Network that contributed patients. Newcastle NIHR Clinical Research Facility: Philip McGrouther, Louise McCormack, Ashley Eglon, Vanessa Ludley. Newcastle Hospitals Pharmacy Department: Ian Campbell, Maria Allen, Lesley Rigdon.
References
Footnotes
Contributors VK: conceived the study, carries the overall responsibility for the full study, the study protocol and the final submitted version of the manuscript; DC: critical review of the manuscript and the revision; HA: wrote the initial draft of this manuscript; NW: statistical input and critical review of manuscript; NH: data management and critical review of manuscript; EM: critical review of manuscript, protocol development; JT: critical review of the protocol; AvW-M: critical review of the manuscript; E-MH: critical review of the manuscript; DS: senior statistical input, protocol development and critical review of the manuscript; GB: critical review of the manuscript; AF: protocol development and critical review of the manuscript; ADS: protocol development and critical review of the manuscript.
Funding The research is supported by an external research grant from AstraZeneca (Funder reference number ISSBRIL0303). The Research was supported/funded by the National Institute for Health Research Newcastle Biomedical Research Centre based at Newcastle Hospitals NHS Foundation Trust and Newcastle University.
Disclaimer The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study has been approved by the North East-Tyne and Wear South Research Ethics Committee (REC 15/NE/0155) and is conducted in accordance with the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.