Article Text

Abstract
Introduction There is currently little evidence for the optimal dosing strategy of four-factor prothrombin complex concentrates (PCC) in vitamin K antagonist (VKA)-related bleeds. The generally accepted dosing strategy is the use of a variable dose calculated using patient-specific characteristics as per manufacturer’s instruction. However, evidence exists that the use of a fixed low dose of 1000 international units of factor IX (IU fIX) might also suffice. Recent studies indicate that in terms of haemostatic effectiveness, the fixed dosing strategy might be even superior to the variable dosing strategy. The PROPER3 (PROthrombin complex concentrate: Prospective Evaluation and Rationalisation, number 3) study aims to confirm the non-inferiority, and explore superiority, in haemostatic effectiveness of the fixed PCC dosing strategy compared with the variable dosing strategy in VKA-related extracranial bleeding emergencies.
Methods and analysis The study is designed as a randomised controlled multicentre non-inferiority trial. Eligibility criteria are an indication for PCC due to VKA-related extracranial bleeding in subjects 18 years of age or older. The control group will receive a variable dose, determined by patient-specific bodyweight and international normalised ratio. The intervention group is dosed a fixed 1000 IU fIX PCC. Primary outcome is the haemostatic effectiveness of both treatments, as defined by the 2016 International Society on Thrombosis and Haemostasis (ISTH) criteria. The sample size is set at 155 patients per treatment arm, requiring 310 patients in total. Non-inferiority on the proportion (risk) difference of the primary outcome will be evaluated using the asymptotic Wald test for non-inferiority. The non-inferiority margin is set at 6%. The primary analysis will be based on the per-protocol population.
Ethics and dissemination Study results will be published in an international journal, communicated to discipline-specific associations and presented at (inter)national meetings and congresses.
Trial registration number EUCTR2014-000392-33; Pre-results.
- anticoagulation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This randomised controlled trial will provide definitive evidence on the effectiveness of the low fixed dose of 1000 international units of factor IX (IU fIX) prothrombin complex concentrate (PCC) compared with the variable dose as per manufacturers instruction, in vitamin K antagonist (VKA)-related bleeding.
The study uses a standardised definition for haemostatic effectiveness as primary outcome, as defined by the International Society on Thrombosis and Haemostasis.
The design is optimised to require minimal study-related effort from treating physicians while exposing participating patients to almost no additional burden.
A deferred consent procedure is used to minimise study-related delay in treatment as PCC for VKA-related bleeding is an emergency situation, therefore allowing capturing of all eligible patients.
A complicating factor might be the wide variety of medical disciplines and physicians involved in treatment of VKA-related bleeds, which might hamper inclusion rates.
Rationale
Vitamin K antagonists (VKAs) are used by millions of users worldwide as oral anticoagulation therapy. Although highly effective, risk of bleeding is an important limitation.1 2 The annual risk of major bleeding is thought to be between 1 and 3 events per 100 patient years,3 4 although recent studies demonstrate incidences ranging from 2.7 to 7.5 events per 100 patient years.5–7 When a major bleed occurs, rapid reversal of VKA is required. For this indication, four-factor prothrombin complex concentrate (PCC) is the agent of choice.8
PCC is a lyophilised concentrate of vitamin K dependent coagulation factors, made from human donor plasma. It has been in routine clinical use in the Netherlands since 1973. However, debate continues about the optimal dosing strategy.9 10 Elements to consider are efficacy regarding cessation of bleeding and also risk of thrombotic complications of PCC as well as speed and ease of administration in an emergency setting and costs.11
The generally accepted dosing strategy is, per manufacturers instruction, dose-calculation based on specific patient characteristics such as bodyweight, international normalised ratio (INR) at presentation and desired target INR (variable dosing strategy). While guidelines advise positively on using PCC, they do not discuss the dosing strategy.12–15
Concurrently, a fixed PCC dosing strategy of 1000 international units of factor IX (IU fIX) is increasingly used in clinical practice. Our pilot study in 2011 demonstrated in a before-and-after design little difference in both INR reduction and clinical outcome when comparing the fixed dosing strategy to the variable dosing strategy.16 A subsequent prospective observational study in extracranial bleeding, in which one hospital using a fixed dosing strategy of 1000 IU fIX was compared with a similar hospital using the variable dosing strategy, demonstrated that the variable dose performed better based on reduction in INR (primary outcome).17 However, when successful clinical outcome was considered, the fixed dose did not perform worse (96% successful clinical outcome in the fixed dose vs 88% in the variable dose).
The main limitation of the study was the lack of randomisation, making this study vulnerable to bias. Although both patient groups and hospitals were similar in all characteristics considered, a difference in baseline risk could not be excluded. Furthermore, the study was not powered on clinical outcome.
In summary, additional evidence is needed to substantiate that the fixed dosing strategy of PCC is at least as clinically effective as a variable dose. We are therefore performing the PROPER3 study (PROthrombin complex concentrate: Prospective Evaluation and Rationalisation, number 3), a randomised controlled multicentre trial to confirm the non-inferiority in clinical efficacy of the fixed dose in a randomised setting.
Objectives
The primary objective of this study is to test whether the clinical outcome, defined as haemostatic effectiveness, of a fixed PCC dose is non-inferior to the variable PCC dose, for VKA-related bleeding in a randomised multicentre setting. The non-inferiority design was selected because if the fixed dose proves to be equally effective, it introduces several advantages over the variable dose. These advantages are easier and quicker administration of an at average lower PCC dose, not requiring dose calculations or specific lab data, a theoretically lower risk of thrombotic complications and as such favouring cost-effectiveness. If non-inferiority is demonstrated, a secondary analysis for superiority is planned.
Secondary objectives are to compare INR as a surrogate endpoint as well as the variables time to PCC administration, total PCC dose, thrombotic complications, length of hospital/ICU stay, additional procedures to reach haemostasis and mortality between the two dosing strategies.
Methods and analysis
Study setting
The study is designed as a randomised controlled non-inferiority trial, comparing the 1000 IU fIX fixed PCC dose strategy to the variable dose PCC to counteract VKAs in extracranial bleeding emergencies. Patient enrolment started in October 2015 and is expected to end in 2019.
The study will mainly take place in the emergency departments of eight large Dutch academic or teaching hospitals. A wide variety of medical disciplines will be involved in the study with the bleeding site determining the treating disciplines. In the Dutch clinical setting, four-factor PCC is the agent of choice for VKA reversal in bleeding emergencies where vitamin K administration alone is not an option. National guidelines advise against the use of plasma or other factor concentrates such as three-factor PCC or activated PCC for this indication.18 Patients enter the study as soon as the treating physician decides to order PCC; the decision itself to use PCC is not part of the study, nor is the PCC supplied by the study.
Eligibility criteria
The study focuses on dose optimisation of PCC in the management of extracranial bleeds in adult VKA users. Eligibility criteria therefore are (1) an indication for PCC due to VKA related bleeding, (2) bleeding is extracranial and (3) subject has to be 18 years or older of age.
Exclusion criteria
Exclusion criteria are: (1) Intracranial bleeding, (2) PCC indication(s) not related to bleeding (eg, urgent reversal in patients without bleeding), (3) PCC indication(s) not related to VKA and (4) previous participation in the study. The exclusion of intracranial bleeds in the current study is based on the lack of pilot data supporting the use of a fixed dose.19
Intervention
Two PCC dosing strategies will be compared in this study. The intervention group will receive a fixed dose of 1000 IU fIX PCC. The control group will receive a variable dose PCC, determined per manufacturer’s instructions. In detail, the variable dose is determined either using bodyweight, baseline INR and target INR (Cofact, Sanquin) or using just bodyweight and baseline INR (Beriplex/KCentra, CSL Behring and Octaplex, Octapharma). See also figure 1 for examples of dose calculation algorithms. The study is open for all PCC brands registered in the Netherlands with participating brands depending on local hospital policy and all brands being four-factor PCC.

PCC dose per manufacturer’s instructions. For Cofact (Sanquin), the variable dose is determined using bodyweight, baseline INR and a choice in a preferred target INR of ≤2.1 (A) or ≤1.5 (B). For Beriplex (CSL Behring), the manufacturer’s instructions only provide a dosing algorithm to reach a target INR≤1.3 (C). For both Cofact and Beriplex, 10 mL corresponds to 250 IU fIX. INR, international normalised ratio; IU fIX, international units of factor IX; PCC, prothrombin complex concentrate.
As mentioned before, the decision to use PCC is at the discretion of the treating physician. All treatment aspects other than the PCC dosing strategy, such as the use of vitamin K or packed red blood cells (PRBCs), should agree with routine clinical practice. This approach was chosen because PCC use and other treatment aspects are highly regulated in the Netherlands by local treatment protocols and national guideline.18 In this way, the study solely intervenes in PCC dose for a proper comparison.
Recruitment and allocation
After deciding to use PCC for a patient fulfilling the eligibility criteria, the treating physician then can randomise the PCC dosing strategy by telephone. Telephone-randomisation was chosen because it requires the least amount of effort from the treating physician. In addition, it is the quickest way to allocate a treatment to the patient, which is crucial to reduce study-related interference in the time to emergency PCC infusion. Finally, using this centralised randomisation maximises allocation concealment pre-randomisation, preventing potential selection bias.
For randomisation GCP-validated ALEA V.15.1 software (FormsVision BV, the Netherlands) is used. Permuted-block randomisation with 1:1 allocation is used, stratified per participating centre. Blinding is not attempted; treatment allocation is open to the treating physician and the patient after randomisation.
Escape medication
If the dose of PCC allocated in the study is deemed not, or not sufficiently, effective, additional PCC dosing is left to the treating physician’s discretion. The treating physician is not limited to a certain timeframe for this decision. When additional PCC is necessary however, the studied treatment for this subject will fail to meet the primary endpoint (ie, no good haemostatic effectiveness reached).
Deferred consent
After a stable condition and situation has been reached, informed consent is sought as soon as possible using a deferred consent procedure. As approved by the ethics committee, a regular informed consent procedure, executed before the study intervention, would harm the patient as the emergency setting of VKA related bleeding does not allow time loss before administration of PCC. As both treatment arms represent a dosing strategy that is routinely used in Dutch clinical practice and local guidelines, randomisation with deferred consent was considered ethical.
Deferred consent is asked from a proxy if the patient is incapable of giving consent personally. For patients or proxies who do not give informed consent, data will not be used. If a randomised patient deceases before being able to give consent, patient data will be used for study purposes to avoid bias.
Primary outcome
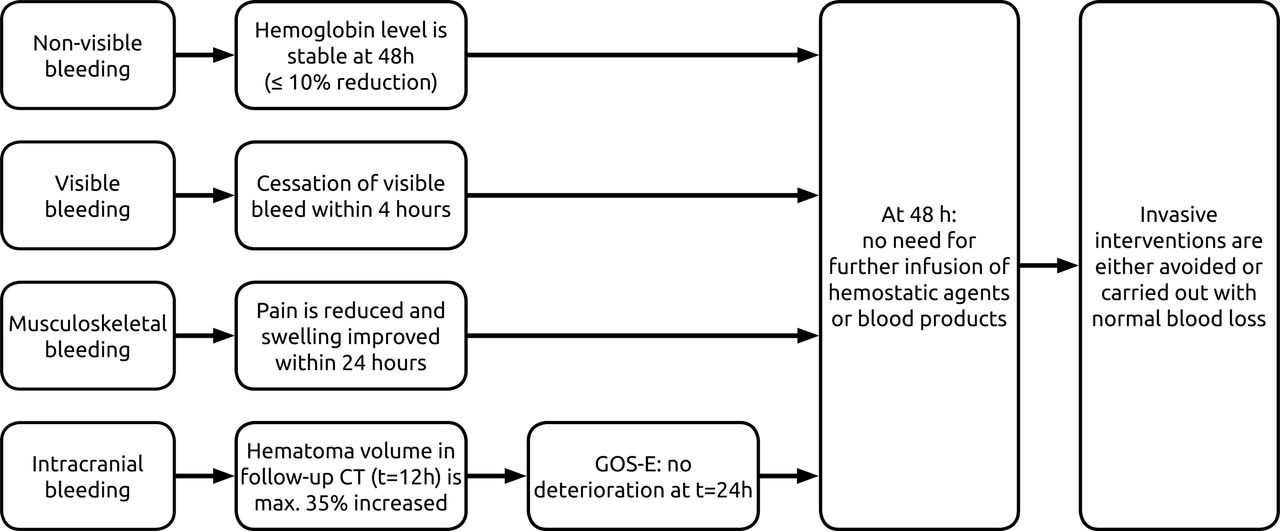
The primary outcome is the proportion of patients with successful clinical outcome, defined as good haemostatic effectiveness, assessed over 24 hours from the end of infusion. Haemostatic effectiveness is defined according to the International Society on Thrombosis and Haemostasis (ISTH) SSC 2016 criteria for assessment of effectiveness of major bleeding management.20 See figure 2 for details of the definition. In summary, haemostasis is considered effective when a visible bleeding is stopped within 4 hours. For a musculoskeletal bleed, there has to be pain relief and improvement in swelling within 24 hours. Effective haemostasis in non-visible bleeds is reached if the haemoglobin level has not decreased more than 10% at 48 hours compared with the level at initial treatment. For all types of bleeds, an additional requirement for haemostasis to be considered effective is that invasive interventions (eg, fasciotomy, endoscopy) were either not necessary or had no bleeding complications.
{kind=link}
{kind=link}
Schematic representation of ISTH proposed definitions for effective haemostasis in management of major bleeding.18 ISTH, International Society on Thrombosis and Haemostasis.
Necessary data for assessment of the haemostatic effectiveness will be collected. With this, data an independent endpoint adjudication board (EAB), blinded to the treatment strategy, will adjudicate the effectiveness of the treatment. The EAB consists of two doctors of internal medicine, who will independently adjudicate the cases at first and in case of disagreement seek consensus.
Secondary outcomes
INR, before and 60 min after the end of PCC administration.
Time between admission to emergency room and start of PCC administration (door-to-needle time).
Repeated dosing and total administered dose of PCC.
Mortality in-hospital and at 30 days after PCC administration.
Thrombotic complications.
Major bleeding complications.
Length of (ICU) hospital stay.
Additional procedures to reach haemostasis.
Sample size
A sample size of 282 patients was calculated to evaluate non-inferiority of the primary endpoint. The study will however include a total of 310 patients to allow for loss to follow-up or patients not evaluable for the primary endpoint. For calculation, first an actual between-group difference of 4% in proportion of patients with effective haemostasis was assumed. This assumption was estimated conservatively from the between group risk difference of 8.3% seen in the highly analogous previous study.17 Second, a non-inferiority margin difference of 6% was deemed clinically acceptable after debate with internationally renowned haematologists and in comparison to previous studies.8 17 PASS software V.11.0.10 (NCSS, Kaysville, Utah, USA) was used to calculate the sample size using the unpooled one-sided Z test, using a required power of 80% and a alpha (one-sided) of 0.025.
Data collection and management
For every included patient, there are two case report forms (CRFs) to complete. The acute phase CRF serves to capture data in the acute situation (ie, around PCC administration) and has to be completed by the treating physician. Only data that are at risk of not being routinely collected are captured by this CRF, such as dose and time of infusion of PCC and other interventions like vitamin K or PRBCs. A paper version was chosen because the emergency situation will not allow the treating physician time to comprehend digital CRF systems. The second CRF collects all other relevant data and is completed by study investigators.
The first CRF will possibly contain patient identifying information and thus will be stored locally in the hospital that included the patient. The second CRF does not contain patient identifying information and is stored centrally at the site of the study coordinator. For study purposes, all data will be handled and processed anonymously. Paper CRF data, excluding any patient identifying information, is subsequently entered in OpenClinica clinical trial data management software V.3.11 (OpenClinica, Waltham, Massachusetts, USA).
Statistical methods
For statistical analysis, we will perform formal hypothesis testing on non-inferiority of the primary outcome, clinical outcome being defined as effective haemostasis, between the fixed and the variable dosing strategy. For this, the asymptotic Wald test for non-inferiority will be used to analyse non-inferiority on the proportion (risk) difference. Asymptotic Wald confidence limits will also be calculated. Analysis will be based on the population that complied with the allocated dosing strategy, that is, the per-protocol population, given the non-inferiority design. A secondary analysis will be performed on all randomised subjects according to the intention-to-treat principle, irrespective of adherence to the allocated dosing strategy.
Non-inferiority of the fixed dosing strategy is demonstrated if the lower limit of the 95% CI of the difference in proportion of patients with effective haemostasis does not exceed (ie, is not lower than) 6%. If non-inferiority is demonstrated, a superiority analysis will follow. Next to 95% CI of the risk difference, Fisher’s exact test will be used to establish superiority.
To determine homogeneity of the observed haemostatic effect over different subgroups of patients, subgroup analyses are planned including patients with high versus low baseline INR (eg, cut-off 4.5) as well as analysis by type of bleeding and severity of bleed, brand of PCC and type of VKA.
Monitoring and auditing
The coordinating investigators will visit study centres approximately once every 3 months, to discuss any issues, check on conduction of the study and discuss inclusion progress. Independent data monitors will audit CRF data yearly.
Ethics and dissemination
The study will be conducted according to the principles of the Declaration of Helsinki (2013) and in accordance with the Dutch Medical Research Involving Human Subjects Act (WMO). As required by the WMO, patients are covered under the insurance for research subjects. The trial is registered in the European Union Clinical Trials Register under EUCTR2014-000392-33 (pre-results).
Study results will be published in an international journal, communicated to discipline-specific associations and presented at (inter)national meetings and congresses. The study was already announced in the Dutch Journal of Medicine (NTvG) of May 2016 to invite centres to participate.21
As of October 2017, 127 patients have been included in 7 participating centres.
Acknowledgments
The authors thank Dr Laurens Nieuwenhuizen and Dr Marieke Kruip for coordinating the PROPER3 study in their hospitals.
References
Footnotes
Contributors RAA, NK, RMVH, NV and KM designed the study and RAA, NK and KM are coordinating the study. LMF, PFY and ETA contributed substantially to improvement of protocol and execution. RAA wrote the protocol manuscript under supervision of KM and NK. All other authors commented on previous versions and contributed to the final version.
Funding The study group received an unrestricted grant from Sanquin Plasma Products BV (Amsterdam, The Netherlands).
Competing interests KM reports grants from Sanquin during the conduct of the study and travel support, speaker fees or consulting fees from Baxter, Bayer, Sanquin, Pfizer, Boehringer Ingelheim, BMS, Aspen and Uniqure outside the submitted work.
Patient consent Not required.
Ethics approval In accordance with Dutch legislation, central study ethics approval was obtained from the medical ethics committee of University Medical Center Groningen (METc UMCG, registered under 2014/285).
Provenance and peer review Not commissioned; externally peer reviewed.