Article Text

Abstract
Introduction This trial proposes to compare the effectiveness and cost-effectiveness of electroacupuncture (EA) plus on-demand gastrocaine with waiting list for EA plus on-demand gastrocaine in providing symptom relief and quality-of-life improvement among patients with functional dyspepsia (FD).
Methods and analysis This is a single-centre, pragmatic, randomised parallel-group, superiority trial comparing the outcomes of (1) EA plus on-demand gastrocaine group and (2) waiting list to EA plus on-demand gastrocaine group. 132 (66/arm) endoscopically confirmed, Helicobacter pylori-negative patients with FD will be recruited. Enrolled patients will respectively be receiving (1) 20 sessions of EA over 10 weeks plus on-demand gastrocaine; or (2) on-demand gastrocaine and being nominated on to a waiting list for EA, which entitles them 20 sessions of EA over 10 weeks after 12 weeks of waiting. The primary outcome will be the between-group difference in proportion of patients achieving adequate relief of symptoms over 12 weeks. The secondary outcomes will include patient-reported change in global symptoms and individual symptoms, Nepean Dyspepsia Index, Nutrient Drink Test, 9-item Patient Health Questionnaire (PHQ9), and 7-item Generalised Anxiety Disorder Scale (GAD7). Adverse events will be assessed formally. Results on direct medical costs and on the EuroQol (EQ-5D) questionnaire will also be used to assess cost-effectiveness. Analysis will follow the intention-to-treat principle using appropriate univariate and multivariate methods. A mixed model analysis taking into account missing data of these outcomes will be performed. Cost-effectiveness analysis will be performed using established approach.
Ethics and dissemination The study is supported by the Health and Medical Research Fund, Government of the Hong Kong Special Administrative Region of China. It has been approved by the Joint Chinese University of Hong Kong – New Territories East Cluster Clinical Research Ethics Committee. Results will be published in peer-reviewed journals and be disseminated in international conference.
Trial registration number ChiCTR-IPC-15007109; Pre-result.
- functional dyspepsia
- acupuncture
- gastrocaine
- randomized controlled trial
- economic evaluation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
We did not use sham control in this trial as we aimed to determine the overall add-on benefit of electroacupuncture, where on-demand gastrocaine is often used as a rescue treatment.
From a pragmatic perspective, non-specific benefits of acupuncture characterised by patient-reported outcomes may be interpreted as clinical effects instead of bias.
To circumvent potential bias caused by the lack of blinding, we have included assessor-blinded objective outcomes (Nutrient Drink Test).
We chose a 12-week follow-up duration, which is in line with current recommendations for trials on patients with functional dyspepsia.
In the future, multicentre trials will provide further evidence on the generalisability of study results.
Introduction
Background and rationale
For patients with uninvestigated dyspepsia who are ≤55 years, current guideline suggests that those with no alarm features undergo test and treatment for Helicobacter pylori when local prevalence is ≥10% (eg, Hong Kong).1 If the patient fails to respond in 4 weeks, current opinion suggests combined use of prokinetics and proton pump inhibitors (PPI) for 8 weeks.2 Oesophagogastroduodenoscopy (OGD) is recommended for those who fail to improve after prokinetics and PPI regimen. A negative finding from OGD suggests the presence of functional dyspepsia (FD), which is defined by the Rome III criteria as ‘the chronic presence of dyspeptic symptoms in the absence of underlying structural or metabolic disease that directly explains the symptoms’.3
FD is divided into two subgroups: postprandial distress syndrome (PDS) and epigastric pain syndrome (EPS). Patients with PDS usually complain of postprandial fullness and early satiation, while those with EPS mainly report epigastric pain and burning.3 It is acknowledged that PDS is more common among Asian patients with FD,4 but it is uncertain whether a separate treatment for patients with PDS and EPS is needed.5
FD is one of the most common gastrointestinal (GI) disorders encountered in clinical practice, with a prevalence of 8% in the Hong Kong population.6 About 50% of patients remain to be symptomatic over 5 years since consultation,7 hence incurring significant treatment cost and loss of productivity. In a Malaysian study, the annual cost of dyspepsia in 1000 population was US$14 816.10 and US$59 282.20, respectively, in rural and urban populations. Among rural and urban adults, respectively, the cost per quality-adjusted life year (QALY) was US$16.30 and US$69.75.8 It is probable that differences in sociodemographic, health service utilisation pattern and clinical factors contribute to cost variation in different settings.9 In South Korea, economic analysis using data from the National Health Insurance Corporation database also indicated that functional GI disorder represents a severe economic burden to the health system.10’
Unfortunately, the effectiveness of PPI and prokinetic for FD is not satisfactory. PPI has a number of needed to treat (NNT) figure of 14.6,11 while a commonly used prokinetics, acotiamide, has an NNT of 16.12 A considerable proportion of FD cases are considered to be refractory to treatment, and some patients opt out from these options due to concerns on their potential adverse effects.13 14
Potential role of electroacupuncture for managing FD
In view of the therapeutic uncertainties of conventional drugs, the potential role of using electroacupuncture (EA) in managing patients with FD can be explored. Multiple therapeutic actions offered by EA may help to manage patients with refractory FD, particularly among those with PDS.15 Existing trials support the effectiveness of EA in managing FD. In a trial conducted by Zeng et al,16 patients with FD receiving EA showed 53.8% improvement in distension and 61.0% improvement in early satiety. In another trial conducted by Ma et al,17 70.7% of patients with FD receiving EA showed marked improvement or had become symptom-free. In a recent network meta-analysis, it is demonstrated that EA is superior to using itopride alone for symptom relief.18
In routine care, on-demand gastrocaine alone is often prescribed as a rescue medication for temporary symptom relief.
Objectives
Primary objective
The primary objective is to determine if EA plus on-demand gastrocaine is superior to waiting list for EA plus on-demand gastrocaine in a proportion of patients with FD of PDS subtype achieving adequate relief of symptoms.
Secondary objectives
The secondary objective is to determine if EA plus on-demand gastrocaine is superior to waiting list for EA plus on-demand gastrocaine in the following outcomes:
patient-reported change in global symptoms and individual symptoms, including postprandial fullness, early satiety, epigastric pain, epigastric burning and postprandial nausea
disease-specific quality of life as measured by the Nepean Dyspepsia Index (NDI)
level of depression as measured by the 9-item Patient Health Questionnaire (PHQ9)
level of postprandial symptoms using Nutrient Drink Test (NDT)
level of anxiety as measured by the 7-item Generalised Anxiety Disorder Scale (GAD7).
The study also aims to determine the cost-effectiveness and safety of EA plus on-demand gastrocaine in managing patients with FD of the PDS subtype, as compared with waiting list plus on-demand gastrocaine.
Adverse events (AEs) from EA and on-demand gastrocaine will be monitored separately using weekly questionnaire.
EuroQol (EQ-5D) questionnaire will be used to estimate QALYs gained.
Direct medical cost associated with the management of FD will be obtained.
Trial design
This is a pragmatic, randomised, controlled, assessor-blinded and data analyst-blinded, single-centre, superiority trial with two parallel groups. Randomisation is performed as block randomisation with random block size, on a 1:1 allocation. This trial is considered to be pragmatic in nature as it aims to investigate whether EA plus on-demand gastrocaine provides more benefits than gastrocaine alone when it is used in routine practice (ie, effectiveness of EA plus gastrocaine). Outcomes that are directly relevant to patients and healthcare professionals are chosen. Overall, this trial is designed to meet the needs of those making a decision (funders and gastroenterology specialists) about referring patients with FD to EA services on top of routine gastrocaine. This pragmatic approach is recommended in a recent consensus statement on comparative effectiveness research for acupuncture.19
Participants, interventions and outcomes
Study setting
This study is recruiting patients from publicly funded gastroenterology specialist clinic, as well as those from the community recruited via newspapers and social media.
Eligibility criteria
All interested subjects will be first assessed for eligibility by a trained, registered Chinese medicine practitioner (RCMP) in accordance with the following criteria, in a face-to-face consultation.
Inclusion criteria
patients who have completed OGD with H. pylori-negative results, or patients who have tested positive for H. pylori but have completed medication course for the eradication of H. pylori
patients with symptoms that fulfil the reference standard for FD PDS, including the presence of either or both of the following symptoms once per week in the past 1 month20:
postprandial fullness
early satiety.
In accordance with the pragmatic approach of this trial, this reference standard is chosen as it reflects presentations of patients with FD in real-world clinical settings, increasing the external validity of future results.
voluntary discontinuation of any conventional pharmacological treatments for their FD 2 weeks prior to enrolment, due to perceived ineffectiveness
Hong Kong permanent resident
18–70 years of age.
Exclusion criteria
Patients who fulfil any of the following criteria would be excluded. Criteria will be assessed through patient history, medical record review or physical examination.
documented diagnosis of oesophageal or gastric disease, including oesophagitis, gastro-oesophageal reflux disease, peptic ulcer, predominant heartburn or acid regurgitation in the past 1 month
current regular user of non-steroidal anti-inflammatory drugs, antidepressants or anxiolytic drugs, defined as daily use in the past 2 months
patients who had received major abdominal surgery
patients who are pregnant
patients who are wearing cardiac pacemaker
patients who are having underlying major physical illness such as malignancy and infections
patients who are using any dose of PPIs or prokinetics 2 weeks prior to enrolment
For patients with coexisting irritable bowel syndrome (IBS) as diagnosed by the Manning criteria, exclusion is applied if patients consider abdominal or bowel symptoms, instead of dyspepsia, as their major complaint. Manning criteria for IBS is positive in this trial when ≥4 of the following symptoms are present in the past month21 22:
visible abdominal distension
pain relieved by a bowel action
more frequent stools with the onset of pain
looser stools with the onset of pain
rectal passage of mucus
a sensation of incomplete evacuation.
Interventions
Intervention group: EA plus on-demand gastrocaine
Patients randomised to the intervention group will receive 20 sessions of EA over 10 weeks (two sessions/week). We encouraged patients to receive treatment according to this schedule, but appointment flexibility is given where patients may opt to complete all treatments in 12 weeks.
A recent randomised trial indicated that EA is superior to itopride in improving symptom scores.17 These improvements can be explained by the deactivating effect of EA on brainstem, anterior cingulate cortex, insula, thalamus and hypothalamus.16 This EA protocol included the acupoints of ST34 (梁丘), ST36 (足三里), ST40 (豐隆) and ST42 (沖陽). Further enhancement of the treatment protocol was performed by consulting two acupuncture experts (WZS and YLJ, both with >30 years of clinical experience) and making reference to literature for additional choices of acupoints.23–26
Combining expert opinions and literature review results, the following points were also added to the treatment protocol: CV12 (中脘), PC6 (内關), BL20 (脾俞) and BL21 (胃俞). In other words, the finalised protocol for this trial has included all of the following points: ST34 (梁丘), ST36 (足三里), ST40 (豐隆), ST42 (沖陽), CV12 (中脘), PC6 (内關), BL20 (脾俞) and BL21 (胃俞). The choice of these points echoes well with Chinese medicine theory, as they are classically used for nourishing qi of spleen, balancing stomach movement, regulating the flow of qi within the liver, as well as in calming the heart and mind. For acupoint locations and standards for locating these points, we followed the WHO Western Pacific Region standard.27 Detailed information can be found in table 1.
Acupoint locations and standards for locating acupoints used in this trial*
Implementation of acupuncture procedures will be standardised and documented according to the Standard for Reporting Interventions in Controlled Trials of Acupuncture requirement28 as follows:
Three RCMPs with full licensure from the Chinese Medicine Council of Hong Kong29 will carry out the procedure. To ensure standardisation of acupuncture treatment, they had copiloted the documented procedure on 10 patients. Treatment compliance will be monitored by patients’ attendance to acupuncture appointments.
RCMP delivering the intervention will insert sterile, single-use filiform acupuncture needles, with a length of 25–40 mm and a diameter of 0.20 mm, with the aid of a guide tube at each of the points, after first disinfecting the skin. The depth of puncture will be made in accordance with the standards of Chinese medicine practice depending on the patient’s body size. The insertion will be followed by a manual bidirectional rotation of the needle sheath in order to produce the sensation known as De qi.
For CV12 and bilateral ST42, the needles will stay for 30 min. For bilateral BL20 and bilateral BL21, further 15 s of bidirectional rotation will be performed after De qi so as to elicit a strong stimulation on these acupoints, and then the needles are removed. Procedures for BL20 and BL21 will be performed at the end of the whole treatment. For the remaining acupoints, EA will be applied bilaterally. Using an EA device, these pairs of acupoints will be subjected to a 30 min 2 Hz continuous wave electrostimulation: PC6–ST34; ST36–ST40. The intensity will be adjusted to produce a muscle twitch that is acceptable to the patient, within the range of 0.5–1.5 mA.
We chose not to apply EA at ST42 and CV12, respectively, due to their proximity with the dorsalis pedis artery and a lack of coupling acupoint. Also, we did not apply electrostimulation at BL20 and BL21 as it is impossible for patients in supine position to receive EA at these two points over the course of treatment. That said, EA remains to be the main modality evaluated in this trial as electrostimulation at the acupoints of ST34, ST36, ST40 and PC6 remained to be much longer than the brief mechanical stimulation at BL20 and BL21. We did not locate any high-quality evidence demonstrating the impact of mechanical acupoint stimulation on the treatment outcome of EA.
For all patients, 12 weeks of on-demand gastrocaine tablets (up to four tablets a day, each tablet contains oxethazaine 5 mg, Al(OH)3 dried gel 126 mg and Mg(OH)2 100 mg) will be provided as a rescue medication. Their use will be recorded by a weekly pill use questionnaire. Patients will be advised not to seek any other conventional or alternative treatments during the trial period. They will be encouraged to seek medical attention at the trial clinic, if needed. In this case, patients will be referred to a doctor who is blinded to treatment assignment. Use of any other additional treatment will be recorded by questionnaire during follow-ups.
Control group: waiting list for EA plus on-demand gastrocaine
For patients randomised to the waiting list plus on-demand gastrocaine group, the same procedure described above would be applied, except for the fact that 20 sessions of EA will not be offered until the completion of all follow-up assessments at 12th week.
In this trial, gastrocaine is a rescue medication to be used in an on-demand basis in both groups. Therefore, it should be considered as a baseline treatment for both groups, not a control treatment. While evidence on antacids for FD is unclear due to a lack of placebo controlled trials, many clinicians and patients worldwide continue to use them.30 Antacids can alleviate acid-related symptoms such as abdominal pain and burning sensations, and may also enhance angiogenesis, bind to bile acid and also reduce peptic activity.31 From the pragmatic perspective of this trial, antacids are a suitable baseline on-demand treatment for a recurrent condition such as FD as they are available without prescriptions and are affordable.32 This choice will allow us to generate real-world evidence on the value of EA in routine practice.
Also, since we are recruiting patients who would like to discontinue conventional pharmacological treatment due to perceived ineffectiveness, the regular use of PPI and prokinetics as controls will not be appropriate for these patients.
To incentivise patients in the control group to stay in the trial, we offered the same EA package as patients in the intervention group once they complete all follow-up assessments. This is an essential step in ensuring low dropout rate in the control group. As shown in figure 1, all measurements are performed prior to the receipt of EA treatment for patients in the control group, and therefore this will not contaminate the results of the trial. It should be emphasised that in Hong Kong, EA is an established treatment modality in Chinese medicine practice, which is under statutory regulation of the Chinese Medicine Council of Hong Kong.33
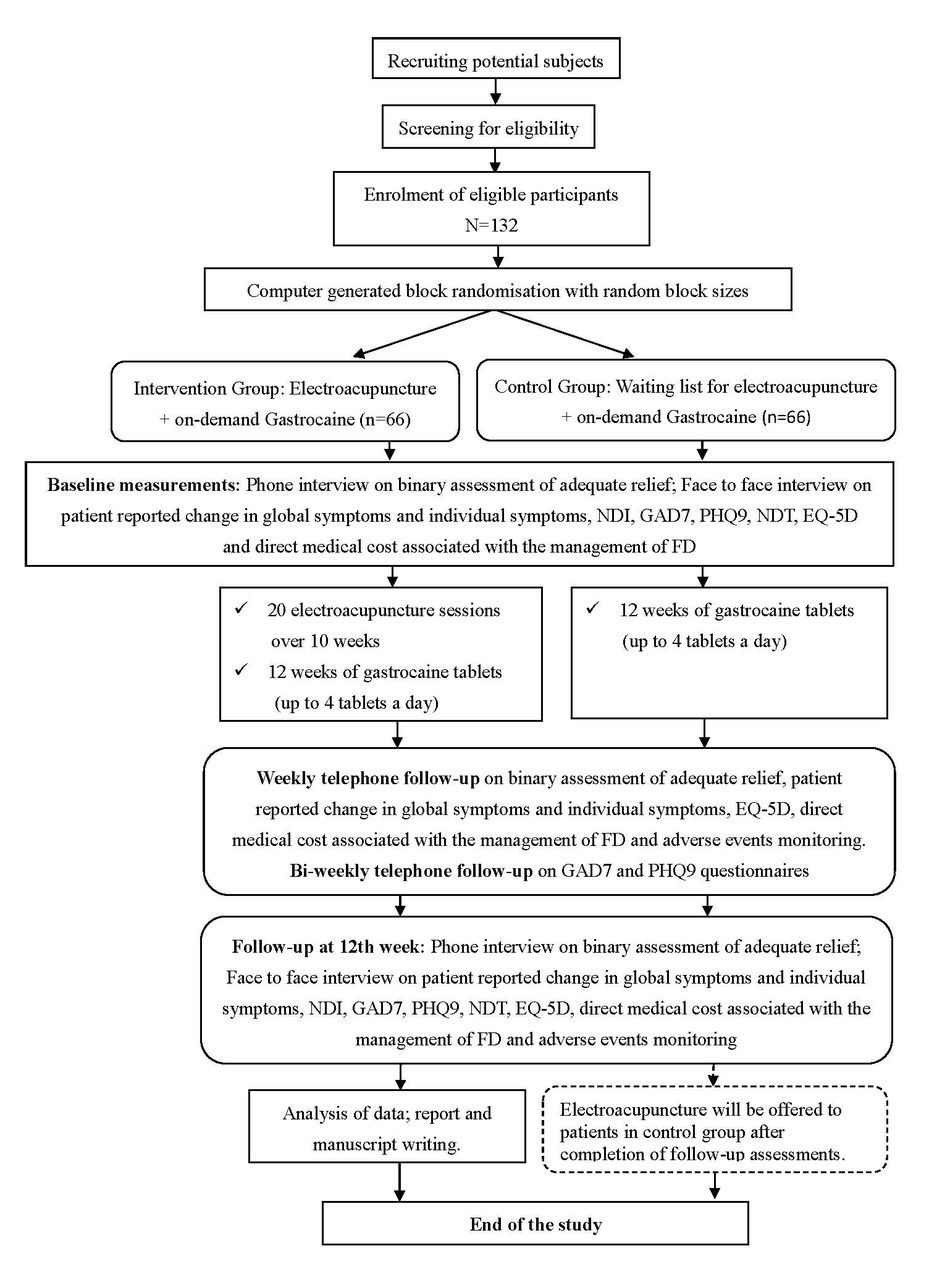
{kind=link}
Flow chart of the proposed trial. FD, functional dyspepsia; GAD7, Patient Health Questionnaire Anxiety Section 7; NDI, Nepean Dyspepsia Index; NDT, Nutrient drink test; PHQ9, Patient Health Questionnaire 9.
Outcomes
Patients in the intervention group will be encouraged to complete all EA treatments in 10 weeks. This will allow us to follow the recommended FD trial follow-up duration of 12 weeks.34 Current literature suggests the use of several distinct approaches in evaluating FD outcomes, including35 (1) primary endpoint: binary assessment of adequate relief; (2) secondary endpoints: patient-reported change in global symptoms; (3) patient-reported change in individual symptoms; (4) disease-specific quality-of-life questionnaires; (5) NDT; and (6) cost-effectiveness outcomes. To ensure consistency in data collection method, all data for all outcomes will be collected via standardised interview by trained, blinded assessors. Details of measurement of these outcomes are provided below.
Binary assessment of adequate relief as primary endpoint
The outcome will be assessed by a weekly question of ‘in the past 7 days, have you had adequate relief of your upper abdominal pain or discomfort? (Yes/No)’. A patient who indicates ‘yes’ for 50% or more at the 3rd–12th weeks of follow-up (ie, over 10 weeks) is considered a responder.36 Answers for the first 2 weeks are not counted as this period is considered an ‘induction period’ for establishing treatment effect.36 These methodological choices are concordant with the recommendations from the Rome III Design of Treatment Trials Committee.37 An improvement of 20% or more over patients in the control group will be considered clinically important.36 This is a recommended primary endpoint for FD trials as it is considered to be responsive, reliable and valid for evaluating improvement.35 36
Patient-reported change in global symptoms and individual symptoms as secondary endpoint
The adequate relief endpoint does not take into account the magnitude of improvement required to achieve a positive response, and it does not reflect worsening of symptoms.35 To overcome this limitation, assessment of symptom change between baseline and the end of the trial is also recommended as an endpoint. Using a 7-point Likert scale (symptoms-free, marked improvement, slight improvement, no improvement, slight deterioration, marked deterioration, severe deterioration), patients are asked to assess the change in global symptoms. Measurements will be made at baseline and at 12th week via face-to-face interview and at weekly intervals in both groups over 12 weeks via phone interview.
For PDS, changes in (1) postprandial fullness and (2) early satiety measured on a 5-point Likert scale (absent, very mild or mild, moderate, severe, very severe) can be measured. A positive response to treatment will be prespecified as improvement by at least one grade in one of the two symptoms, with no deterioration on the other one.34 35 An improvement of 20% or more over patients in the control group will be considered clinically important.36 Data will be collected via face-to-face interview at baseline and at the end of the trial (ie, 12th week) and at weekly intervals in both groups over 12 weeks via phone interview. Changes in epigastric pain, epigastric burning and postprandial nausea will also be recorded and interpreted in a similar fashion, but they will be regarded as supplementary endpoints.
Change in disease-specific quality-of-life questionnaire scorings as secondary endpoint
Assessment of adequate relief and individual symptom change alone may not reflect wider impact potentially brought by acupuncture. To address this gap, the NDI will be used as a secondary outcome. It is a psychometrically sound, responsive questionnaire for measuring improvement in quality of life among patients with FD.34 35 A Chinese version of the NDI has been validated, with satisfactory internal consistency, validity and reliability.38 A change of 10 points on the total NDI scale indicates clinically important change on patients’ health status.39 For NDI, measurements will be made at baseline and at the end of the trial (ie, 12th week) via face-to-face interview.
As depression and anxiety are common comorbidities of FD,40 changes in depressive symptoms will be assessed by PHQ9. For changes in anxiety symptoms, GAD7 will be used.41 For PHQ9, a Chinese version has been validated,42 and a five-point difference on the scale indicates clinically important changes.43 Validated Chinese version for GAD7 is also available,44 and the minimally important change is also five points.19 For PHQ9 and GAD7, measurement will be made at baseline and at 12th week via face-to-face interview and at biweekly intervals in both groups over 12 weeks via phone interview.
We have chosen PHQ9 and GAD7 instead of the Hospital Anxiety and Depression (HAD) questionnaire as the underlying structure of this instrument is inconsistent across samples and is highly dependent on the statistical methods used to establish that structure. This implies that the HAD is not a dependable means of differentiating anxiety and depression for the purposes of assessing the absolute or relative levels of these variables.45 Recently, there has been call for abandonment of HAD.46 47
NDT results as secondary outcome
Current literature on FD trial design recommends the use of NDT as an objective outcome to assess the impact of treatment on postprandial symptoms.34 The maximum tolerated volume as well as the extent of symptom aggregation (nausea, fullness, bloating and pain) 30 min after the test will be measured. Standardised NDT procedures, normal values for maximum tolerated volume and symptom severity have been published.48 Patients’ performance at baseline and at 12th week will be compared against these standards. The NDT will be performed at baseline and at the end of the trial according to published procedures.48
Cost-effectiveness outcome
QALYs gained will be estimated for each patient using the EuroQol (EQ-5D) questionnaire. EQ-5D is a valid and reliable instrument for measuring quality of life of patients with FD.49 The EQ-5D scores and direct medical costs associated with the management of FD will be obtained at baseline and at 12th week via face-to-face interview and at weekly intervals in both groups over 12 weeks via phone interview.
Participants’ timeline
Flow and timeline of the proposed trial can be found in figure 1.
Sample size estimation
The sample size was calculated on the basis of the following hypothesised change in primary outcome (binary assessment of adequate relief; for details, please see the Binary assessment of adequate relief as primary endpoint section): intervention group is superior to control group by 20% in providing adequate relief of symptoms in patients with FD of PDS subtype. Investigation of the natural history of FD demonstrates that 60% of patients will remain symptomatic over a long period of time.50 Conservatively, it can be expected that only 10% of patients in the control group would improve, given the fact that their condition is of refractory nature and no active treatments were provided. Taking into account expert consensus-based minimally important clinical difference of 20%,36 it is expected that 30% of patients in the intervention group would improve if additional EAs were deemed valuable.
Our estimation of an improvement of 30% from baseline among patients in the intervention group should be considered conservative, as results from previous published trials using EA for FD showed much larger effect sizes. In a trial conducted by Zeng et al,16 patients with FD receiving EA showed 53.8% improvement in distension and 61.0% improvement in early satiety. In another trial conducted by Ma et al,17 70.7% of patients with FD receiving EA showed marked improvement or had become symptom-free.
To achieve 80% power, with significance level at 5%, 118 patients are required. Assuming that refusal of follow-up will occur in 10% of patients, a minimum of 66 patients are required in each arm of the study. The total number of patients to be enrolled will therefore be 132. The power calculation was based on two independent proportion tests, using PASS 13 (V.13.0.13; NCSS Statistical Software).
Assignment of interventions
Sequence generation
Blocked randomisation will be used to allocate patients to the two groups at 1:1 ratio, with random block sizes.51 Sequence will be generated by the Random Allocation Software.52
Allocation: concealment mechanism
Adhering to published method, random sequence will be concealed using the sequentially numbered, opaque sealed envelopes (SNOSE) procedure.53
Allocation: implementation
These SNOSEs will be kept by a person not involved in the care or evaluation of patients, or in the data analysis. Investigators enrolling patients will have no access to SNOSE. Treatment allocation process will start when the investigator calls the personnel keeping the SNOSE. The computer database will be designed in such a way that treatment allocation cannot be changed after randomisation.
Blinding
Blinding of data analysts and outcome assessors will be ensured. The assessors will receive rigorous training in standardised data collection procedures. Data entry personnel external to the research team will be employed to perform data entry such that the data analysts will analyse data without the need to refer to allocation information. Professor Ben Yip and Dr Irene Wu from The Chinese University of Hong Kong, who are not involved in the conduct of this trial, will perform the data analysis.
Given the nature of EA intervention, blinding of investigators and patients is unsatisfactory. They will be strongly discouraged from disclosing patients’/own allocation status to data analysts and outcome assessors. To circumvent the lack of patient and investigator blinding, we have proposed the use of NDT as an objective outcome. It is acknowledged that the impact of lack of blinding is minimal in the measurement of objective outcomes.54
Data management and analysis
Data management
All hardcopy patient report forms, in numerical order, will be kept in locked cabinets. Access to the study data will be restricted. Electronic backup of all data will be created and retained in a separate secure location on a weekly basis. All data will be entered electronically by data entry personnel not involved in the trial. At the completion of data entry, a random subset will be selected for quality control. Data from selected report forms will be compared against the entered data. Data integrity will be enforced through further mechanisms including referential data rules, valid values, range checks and consistency checks against data stored in databases. All modifications to data written to the database will be documented. Interim auditing of the trial was performed in January 2017.
Statistical methods and cost-effectiveness analysis
The main analysis will adhere to the intention-to-treat principle, in which all randomised patients will be included. For safety data (see reporting of harms section) analysis, only patients who have attended at least one session of EA treatment will be included. Demographic and baseline characteristics will be compared between intervention and control groups descriptively. Relative risk reduction with 95%CI will also be calculated for binary outcomes. Continuous outcomes will be reported as means and SD, or medians and IQR, as appropriate. Categorical variables will be presented as frequencies and percentages. Χ2 test will be used for the primary endpoint and safety data. Student’s t-test or χ2 test will be performed for the secondary outcomes. All P values and 95% CIs are two-sided, with a P value of less than 0.05 considered to indicate statistical significance. The Bonferroni method will be used to appropriately adjust the overall level of significance for multiple outcomes.
Baseline characteristics will be adjusted using multivariate analyses. Logistic regression will be used for binary outcomes, and linear regression will be used for continuous outcome. Model assumption and goodness of fit will be assessed by examining the residuals. Taking into account missing data due to attrition, mixed model analysis will also be performed.55
For cost-effectiveness analysis, a standard multiplicative model will be used to estimate QALYs by the area under linear interpolation of the EQ-5D score trajectory for each individual using weekly intervals.56 The QALYs gained in each study arm will be presented as mean with SD. In addition, patients’ direct medical costs will be obtained at baseline via face-to-face interview and at weekly intervals in both groups over 12 weeks via phone interview.
Reporting of harms
AEs will be monitored by a questionnaire given to all participants at weekly intervals in both groups over 12 weeks via phone interview. For the intervention group, the AE questionnaire will include the following questions57:
Have you experienced any symptoms which you think might be caused by your electroacupuncture treatment? (yes/no).
If yes, please describe.
How likely do you think it is the acupuncture procedure that caused these symptoms? (definitely/probably/possibly/unlikely).
Have you experienced any symptoms which you think might be caused by gastrocaine? (yes/no).
If yes, please describe.
How likely do you think it is gastrocaine that caused these symptoms? (definitely/probably/possibly/unlikely).
For the control group, only questions 4–6 will be asked. Causality between EA/gastrocaine and AE will follow the assessment scheme devised by Vickers et al.57 Patients will also be given a telephone number to call if they have any enquiries on AE during the treatment period.
Ethical consideration
Consent
The principal investigator (VCHC), who is a RCMP, will introduce the trials to patients orally with aid from a PowerPoint presentation in a face-to-face appointment. An information sheet will also be given to the patients. Informed patients will then be given time to discuss possible enrolment with the principal investigator. For eligible patients who are willing to join, formal written consents were obtained.
Confidentiality
All hardcopies of study-related information will be stored securely in a location with restricted access. In electronic database files, depersonalised codes will be created to replace participants’ identifying information. All electronic files will be encrypted and password-protected. Only authorised research personnel will be able to access hardcopy and electronic data sets.
Acknowledgments
The authors would like to thank the following personnel for supporting the implementation of this trial: Robin ST Ho, William KW Cheung, Jasmine TM Chow, Irene XY Wu and Benjamin HK Yip.
References
Footnotes
WZS, YLJ, SSH, WLL and KCL contributed equally.
Contributors Study concept and design: VCHC. Drafting of the manuscript: VCHC, CHLW. Critical revision of the manuscript for important intellectual content: VCHC, SYSW, JCYW. Administrative, technical or material support: CHLW, JYLC, WZS, YLJ, SSH, WLL, KCL. Figure preparation: CHLW. All authors reviewed the manuscript, approved all the contents and agreed on the submission.
Funding This trial is funded by the Health and Medical Research Fund, Hong Kong SAR Government (ref no: 12130211).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethical approval has been obtained from the Joint Chinese University of Hong Kong – New Territories East Cluster Clinical Research Ethics Committee (ref no: 2014.552 T). The protocol, informed consent form, participant recruitment materials and other request documents were reviewed and approved by this committee before implementation.
Provenance and peer review Not commissioned; externally peer reviewed.